

Streptococcus agalactiae clones infecting humans were selected and fixed through the extensive use of tetracycline
- PMID: 25088811
- PMCID: PMC4538795
- DOI: 10.1038/ncomms5544
Streptococcus agalactiae clones infecting humans were selected and fixed through the extensive use of tetracycline
Erratum in
-
Corrigendum: Streptococcus agalactiae clones infecting humans were selected and fixed through the extensive use of tetracycline.Nat Commun. 2015 Jan 28;6:6108. doi: 10.1038/ncomms7108. Nat Commun. 2015. PMID: 25629801 Free PMC article. No abstract available.
Abstract
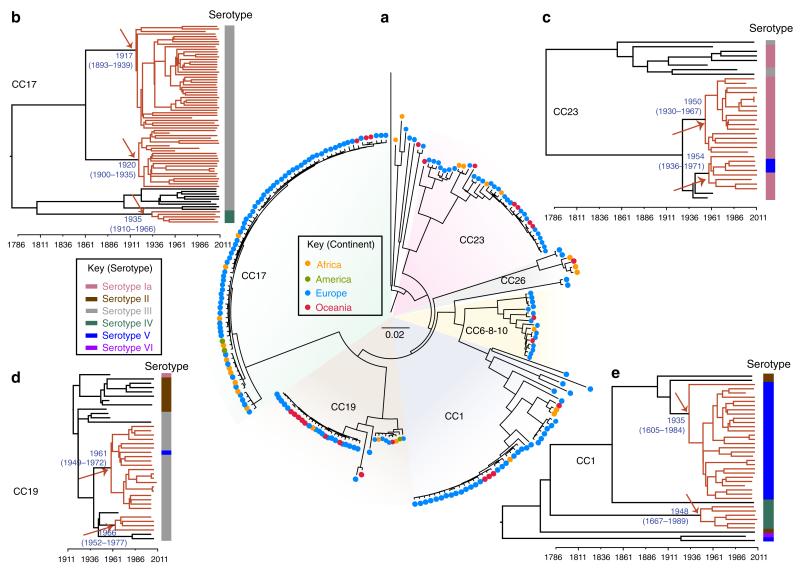
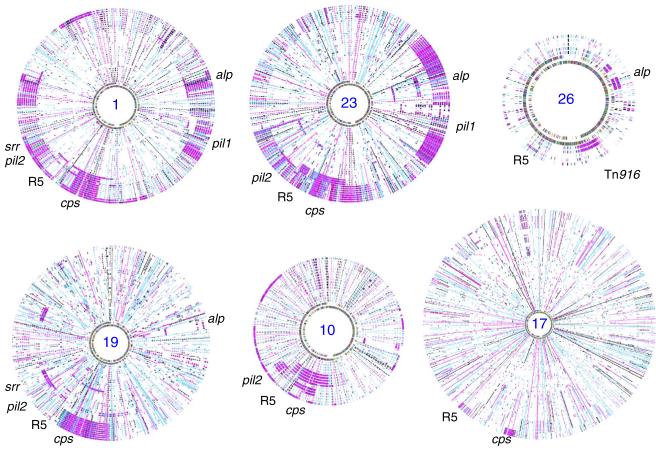
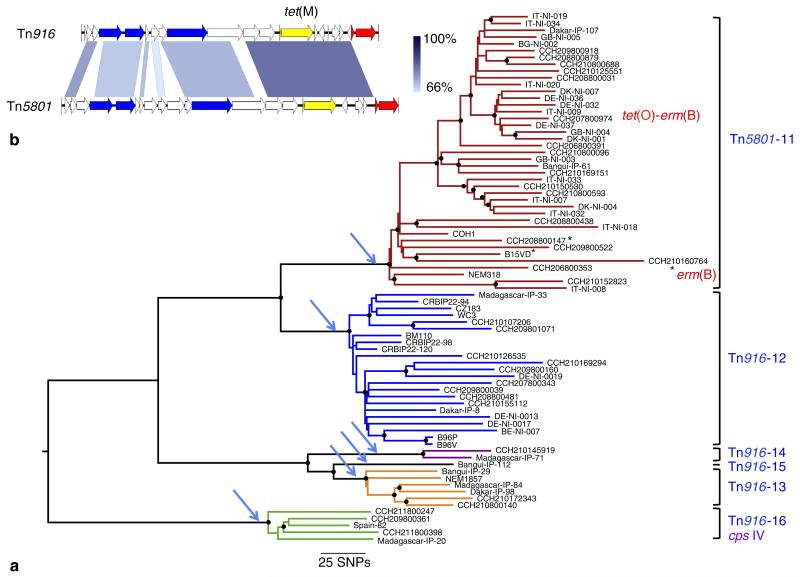
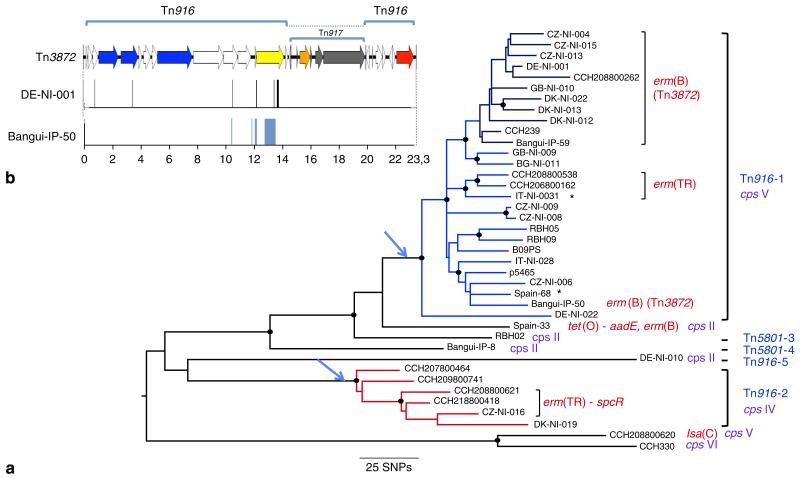
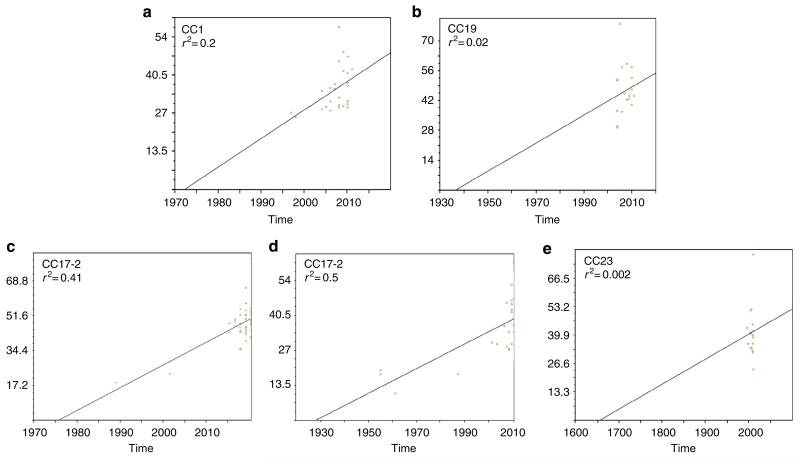
Streptococcus agalactiae (Group B Streptococcus, GBS) is a commensal of the digestive and genitourinary tracts of humans that emerged as the leading cause of bacterial neonatal infections in Europe and North America during the 1960s. Due to the lack of epidemiological and genomic data, the reasons for this emergence are unknown. Here we show by comparative genome analysis and phylogenetic reconstruction of 229 isolates that the rise of human GBS infections corresponds to the selection and worldwide dissemination of only a few clones. The parallel expansion of the clones is preceded by the insertion of integrative and conjugative elements conferring tetracycline resistance (TcR). Thus, we propose that the use of tetracycline from 1948 onwards led in humans to the complete replacement of a diverse GBS population by only few TcR clones particularly well adapted to their host, causing the observed emergence of GBS diseases in neonates.
Figures
References
-
- Edwards MS, Baker CJ. Group B streptococcal infections. In: Klein JO, Remington JS, editors. Infectious Diseases of the Fetus and Newborn Infant. Saunders; 2005.
-
- Edmond KM, et al. Group B streptococcal disease in infants aged younger than 3 months: systematic review and meta-analysis. Lancet. 2012;379:547–556. - PubMed
-
- Eickhoff TC, Klein JO, Daly AK, Ingall D, Finland M. Neonatal sepsis and other infections due to group B beta-hemolytic streptococci. N. Engl. J. Med. 1964;271:1221–1228. - PubMed
-
- Kexel G, Schoenbohm S. Streptococcus agalactiae as the causative agent in infantile meningitis. Dtsch. Med. Wochenschr. 1965;90:258–261. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
