

Late-Onset Glycogen Storage Disease Type II (Pompe's Disease) with a Novel Mutation: A Malaysian Experience
- PMID: 25093132
- PMCID: PMC4100255
- DOI: 10.1155/2014/926510
Late-Onset Glycogen Storage Disease Type II (Pompe's Disease) with a Novel Mutation: A Malaysian Experience
Abstract
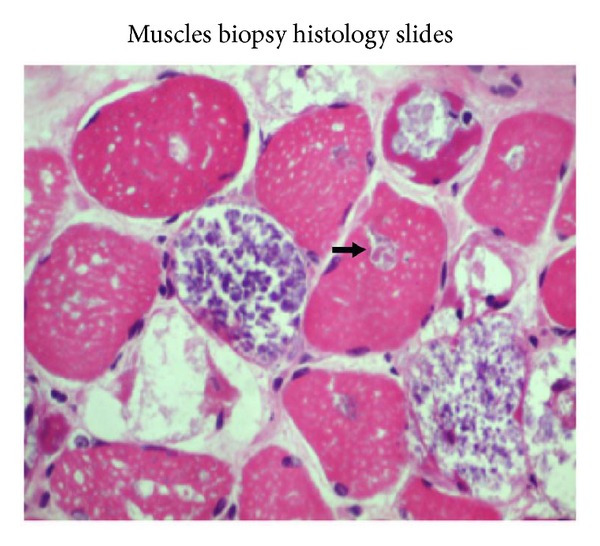
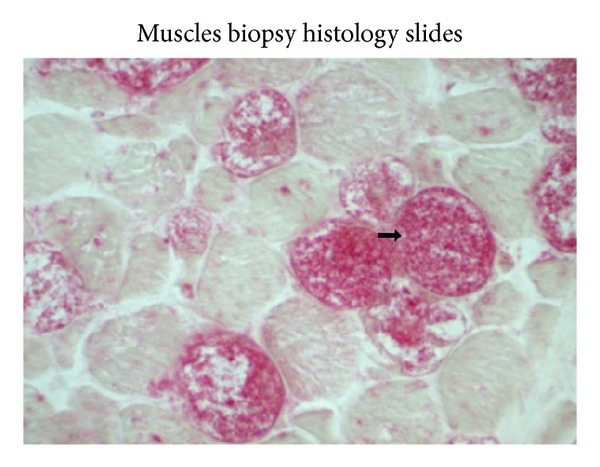
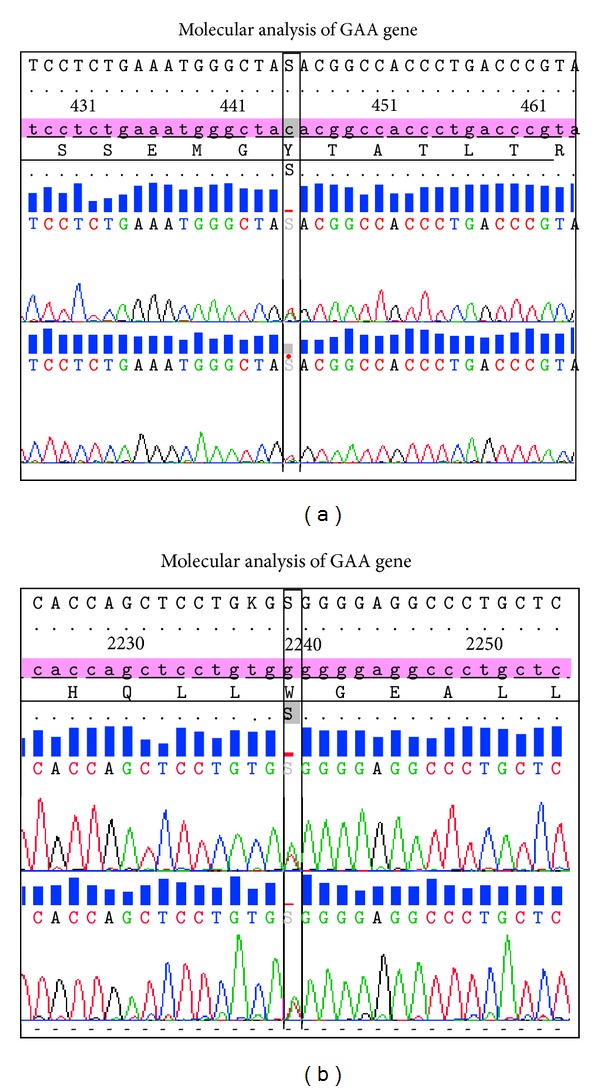
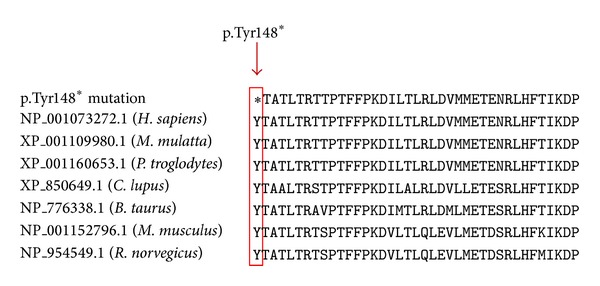
Pompe's disease (acid maltase deficiency, glycogen storage disease type II) is an autosomal recessive disorder caused by a deficiency of lysosomal acid α-1,4-glucosidase, resulting in excessive accumulation of glycogen in the lysosomes and cytoplasm of all tissues, most notably in skeletal muscles. We present a case of adult-onset Pompe's disease with progressive proximal muscles weakness over 5 years and respiratory failure on admission, requiring prolonged mechanical ventilation. Electromyography showed evidence of myopathic process with small amplitudes, polyphasic motor unit action potentials, and presence of pseudomyotonic discharges. Muscle biopsy showed glycogen-containing vacuoles in the muscle fibers consistent with glycogen storage disease. Genetic analysis revealed two compound heterozygous mutations at c.444C>G (p.Tyr148∗) in exon 2 and c.2238G>C (p.Trp746Cys) in exon 16, with the former being a novel mutation. This mutation has not been reported before, to our knowledge. The patient was treated with high protein diet during the admission and subsequently showed good clinical response to enzyme replacement therapy with survival now to the eighth year. Conclusion. In patients with late-onset adult Pompe's disease, careful evaluation and early identification of the disease and its treatment with high protein diet and enzyme replacement therapy improve muscle function and have beneficial impact on long term survival.
Figures
References
-
- Leslie N, Tinkle BT. Glycogen storage disease type II (pompe disease) GeneReviews. 2007
-
- Hirschhorn R, Reuser AJJ. Glycogen storage disease type II: (acid maltase) deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Vogelstein B, editors. The Online Metabolic and Molecular Bases of Inherited Disease (OMMBID) chapter 135. New York, NY, USA: McGraw-Hill; 2013.
-
- Yang CC, Chien YH, Lee NC, et al. Rapid progressive course of later-onset Pompe disease in Chinese patients. Molecular Genetics and Metabolism. 2011;104(3):284–288. - PubMed
-
- Qiu J. J., Wei M, Zhang WM, Qiu JJ, Meng Y, Qiu ZQ. Clinical and molecular genetic study on two patients of the juvenile form of Pompe disease in China. Zhonghua er ke za zhi. 2007;45(10):760–764. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
