

FAK in cancer: mechanistic findings and clinical applications
- PMID: 25098269
- PMCID: PMC4365862
- DOI: 10.1038/nrc3792
FAK in cancer: mechanistic findings and clinical applications
Abstract
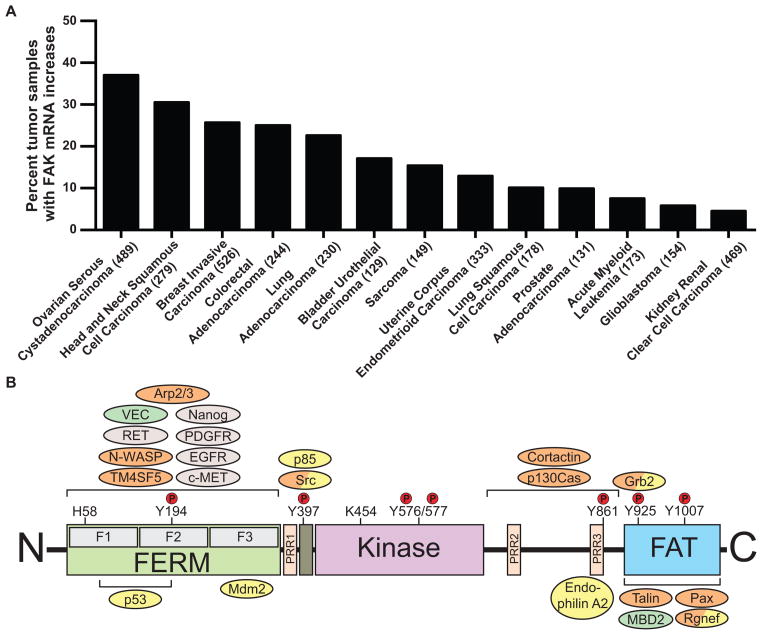
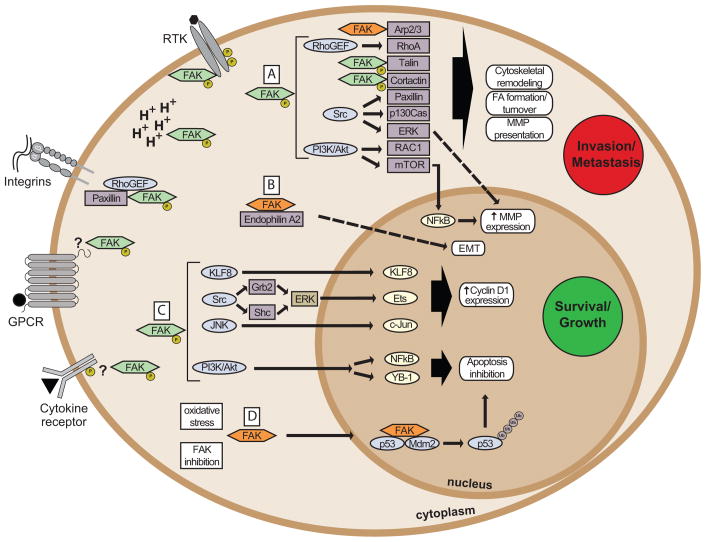
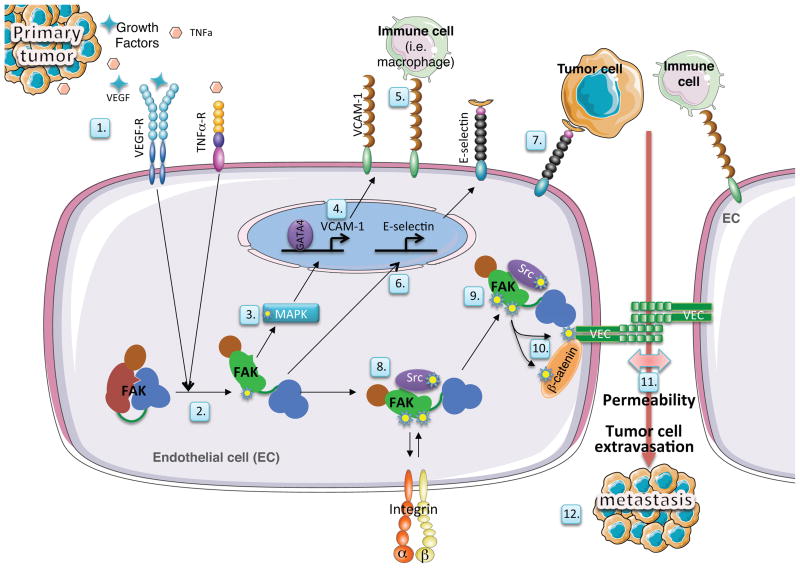
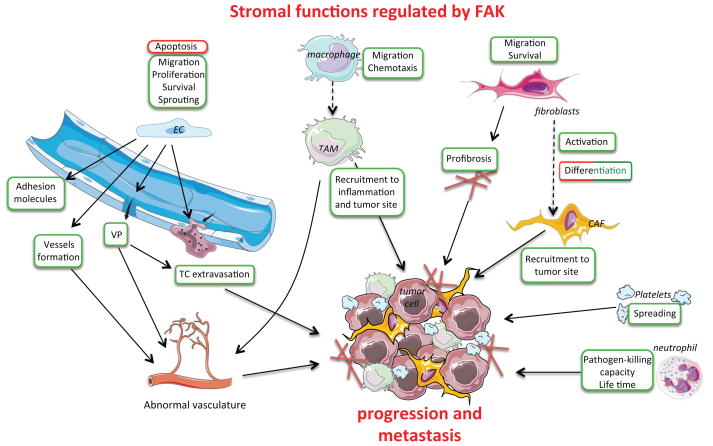
Focal adhesion kinase (FAK) is a cytoplasmic protein tyrosine kinase that is overexpressed and activated in several advanced-stage solid cancers. FAK promotes tumour progression and metastasis through effects on cancer cells, as well as stromal cells of the tumour microenvironment. The kinase-dependent and kinase-independent functions of FAK control cell movement, invasion, survival, gene expression and cancer stem cell self-renewal. Small molecule FAK inhibitors decrease tumour growth and metastasis in several preclinical models and have initial clinical activity in patients with limited adverse events. In this Review, we discuss FAK signalling effects on both tumour and stromal cell biology that provide rationale and support for future therapeutic opportunities.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116:1409–16. - PubMed
-
- Schaller MD. Cellular functions of FAK kinases: insight into molecular mechanisms and novel functions. J Cell Sci. 2010;123:1007–13. - PubMed
-
- Zhao J, Guan JL. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009;28:35–49. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
