

Innate immunity pathways regulate the nephropathy gene Apolipoprotein L1
- PMID: 25100047
- PMCID: PMC4312530
- DOI: 10.1038/ki.2014.270
Innate immunity pathways regulate the nephropathy gene Apolipoprotein L1
Abstract
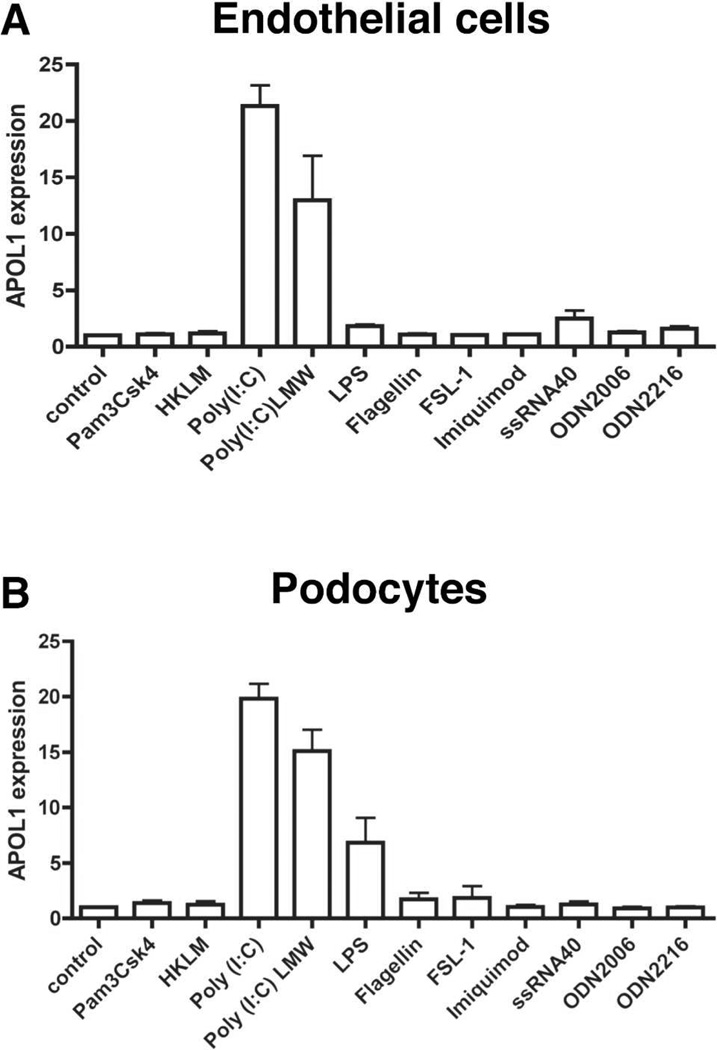
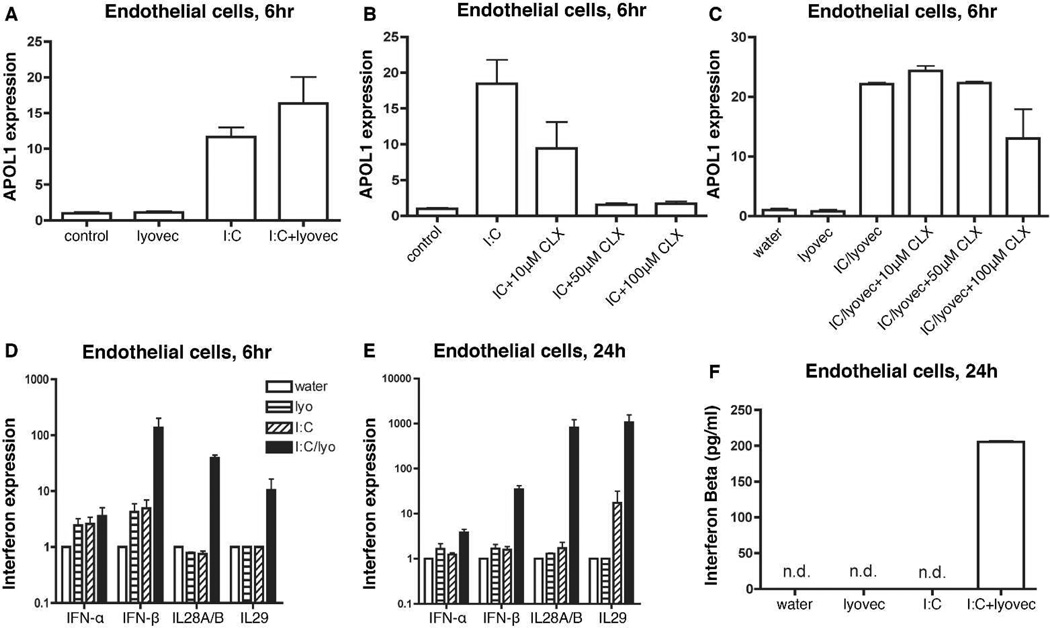
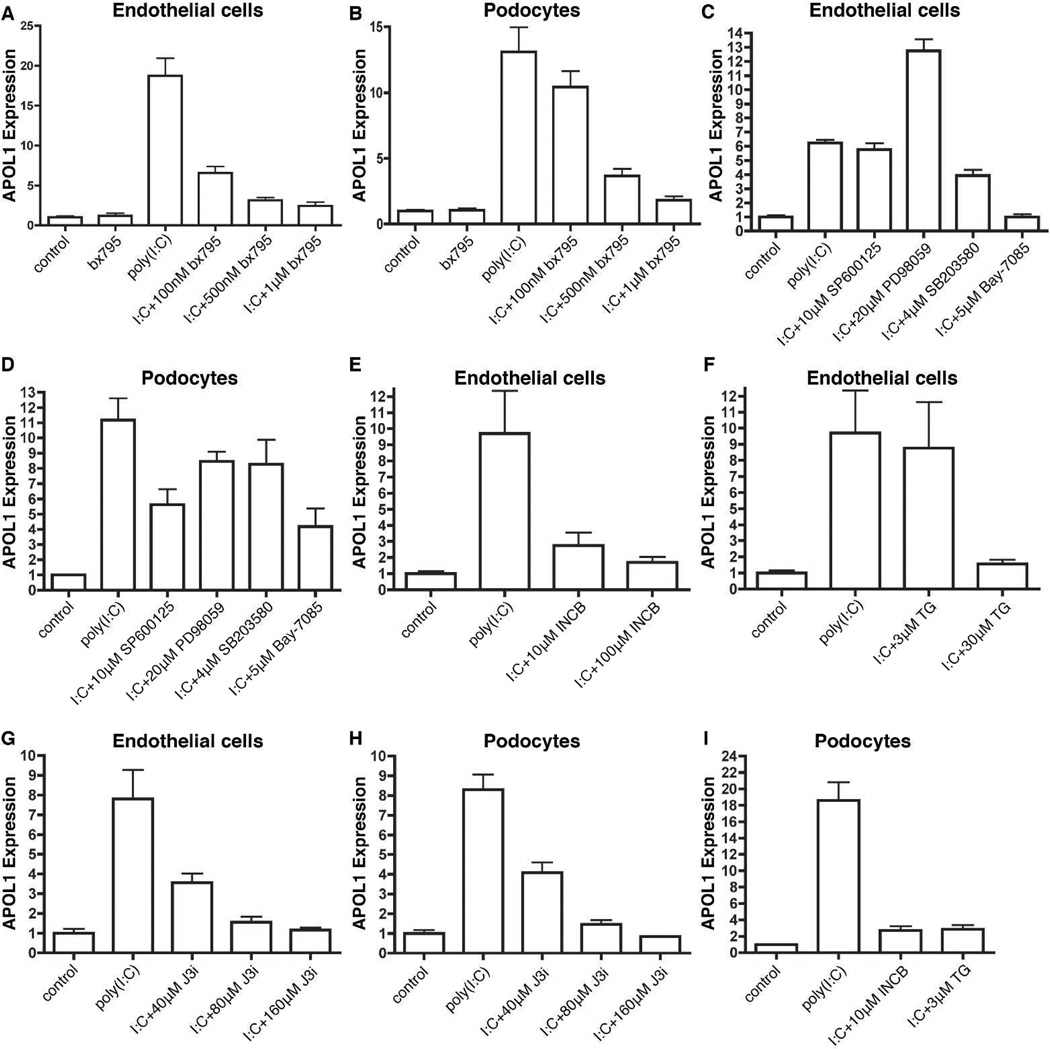
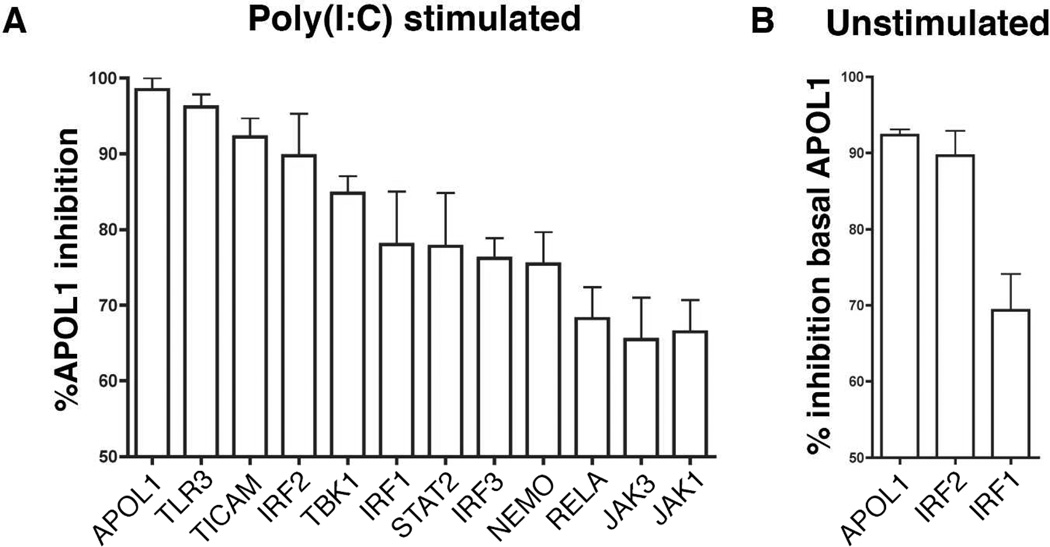
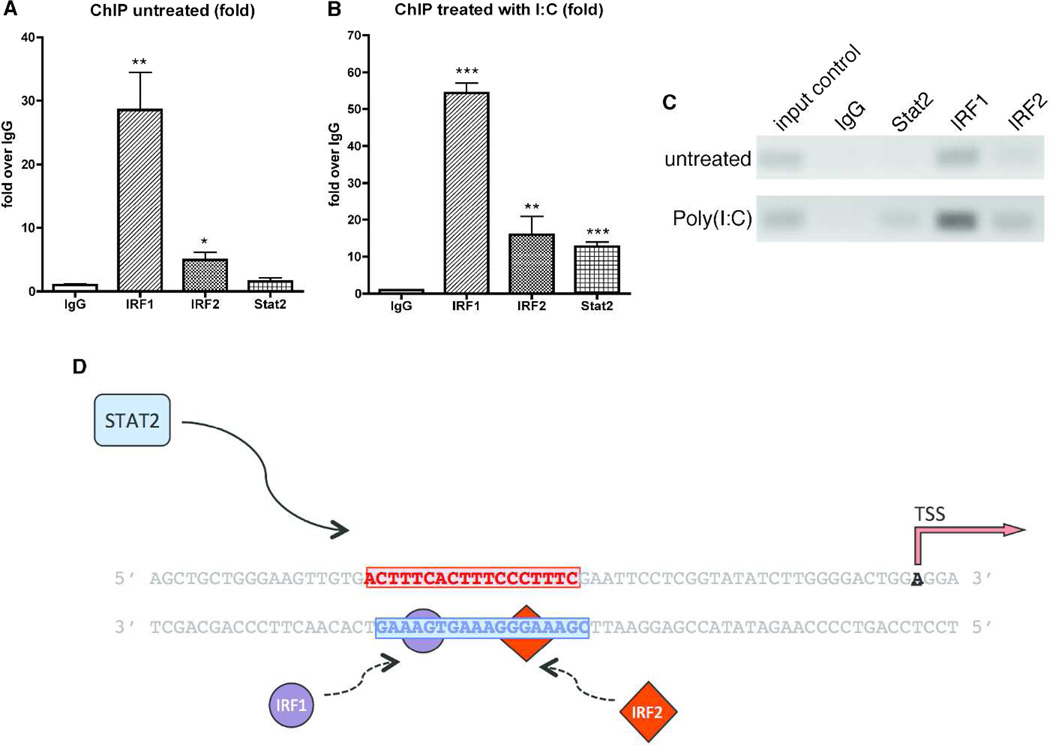
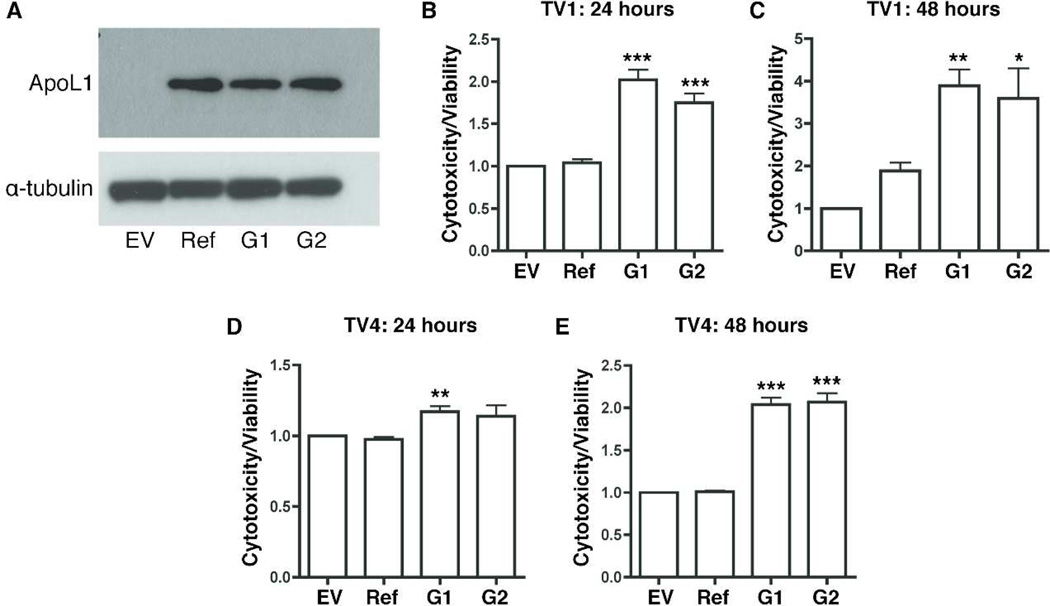
Apolipoprotein L1 (APOL1) risk variants greatly elevate the risk of kidney disease in African Americans. Here we report a cohort of patients who developed collapsing focal segmental glomerulosclerosis while receiving therapeutic interferon, all of whom carried the APOL1 high-risk genotype. This finding raised the possibility that interferons and the molecular pattern recognition receptors that stimulate interferon production may contribute to APOL1-associated kidney disease. In cell culture, interferons and Toll-like receptor (TLR) agonists increased APOL1 expression by up to 200-fold, in some cases with the appearance of transcripts not detected under basal conditions. PolyI:C, a double-stranded RNA TLR3 agonist, increased APOL1 expression by upregulating interferons directly or through an interferon-independent, IFN-regulatory factor 3 (IRF3)-dependent pathway. Using pharmacological inhibitors, small hairpin RNA knockdown, and chromatin immunoprecipitation, we found that the interferon-independent TLR3 pathway relied on signaling through TBK1, NF-κB, and Jak kinases, and on binding of IRF1, IRF2, and STAT2 at the APOL1 transcription start site. We also demonstrate that overexpression of the APOL1 risk variants is more injurious to cells than overexpression of the wild-type APOL1 protein. Our study illustrates that antiviral pathways may be important inducers of kidney disease in individuals with the APOL1 high-risk genotype and identifies potential targets for prevention or treatment.
Conflict of interest statement
David Friedman and Martin Pollak are co-inventors on patents filed by Beth Israel Deaconess Medical Center related to APOL1 diagnostics and therapeutics.
Figures

Comment in
-
Glomerular disease: innate immunity-APOL1 interaction.Nat Rev Nephrol. 2014 Oct;10(10):543. doi: 10.1038/nrneph.2014.158. Epub 2014 Aug 26. Nat Rev Nephrol. 2014. PMID: 25157835 No abstract available.
-
Immunity unmasks APOL1 in collapsing glomerulopathy.Kidney Int. 2015 Feb;87(2):270-2. doi: 10.1038/ki.2014.325. Kidney Int. 2015. PMID: 25635718
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
