

Coronaviruses resistant to a 3C-like protease inhibitor are attenuated for replication and pathogenesis, revealing a low genetic barrier but high fitness cost of resistance
- PMID: 25100843
- PMCID: PMC4178758
- DOI: 10.1128/JVI.01528-14
Coronaviruses resistant to a 3C-like protease inhibitor are attenuated for replication and pathogenesis, revealing a low genetic barrier but high fitness cost of resistance
Abstract
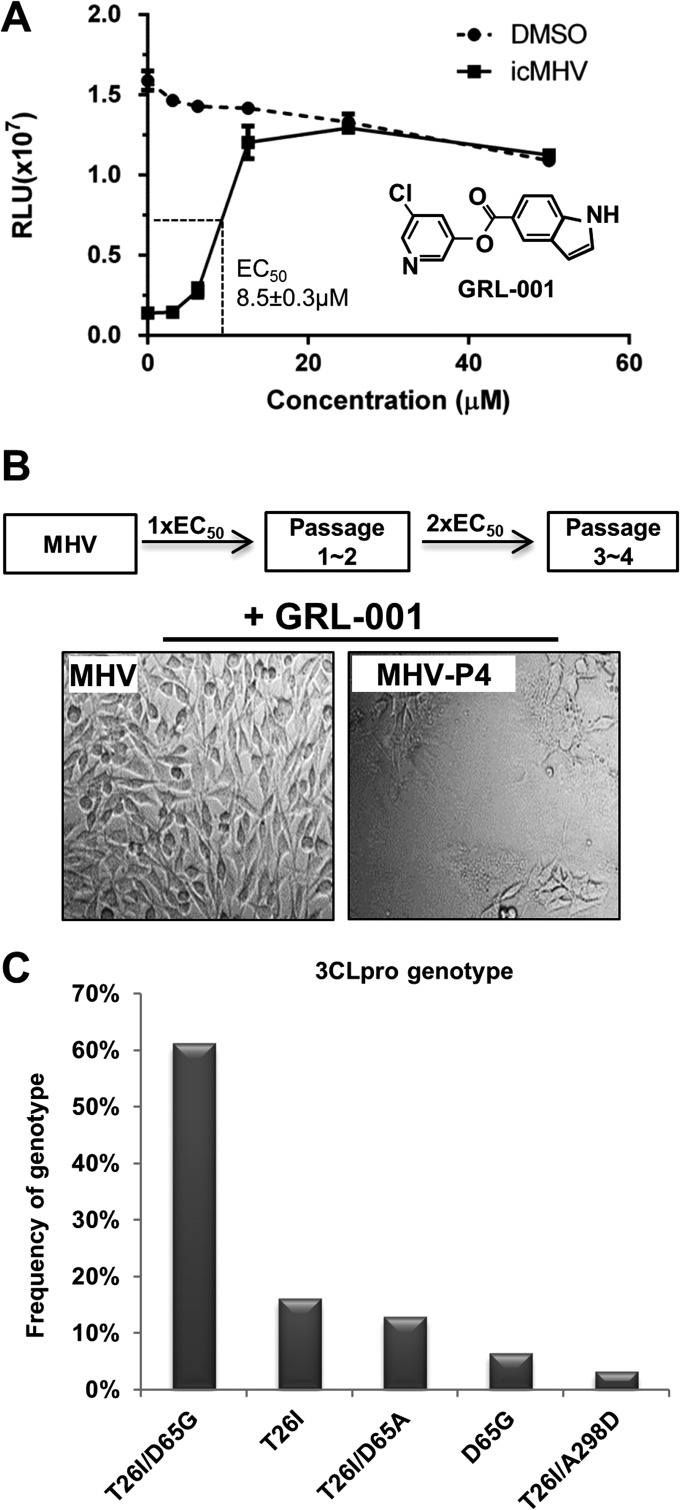
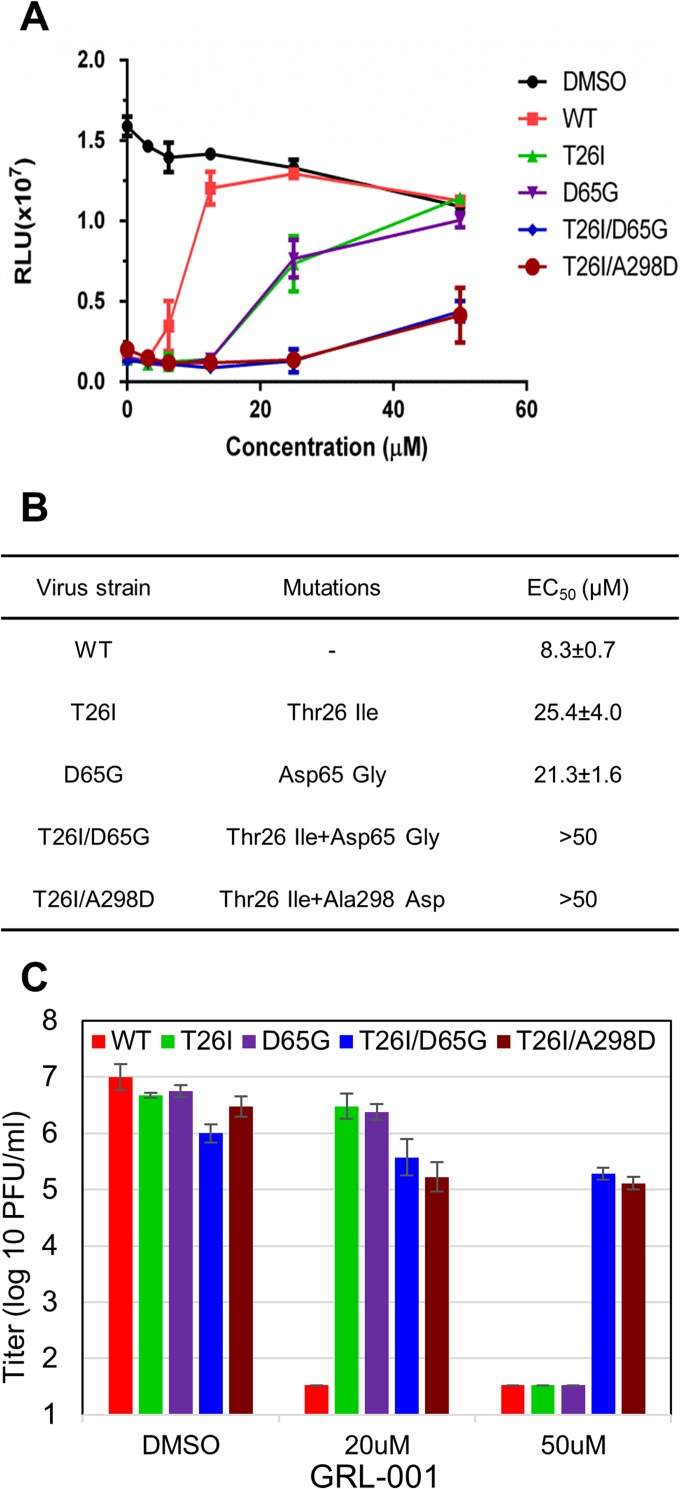
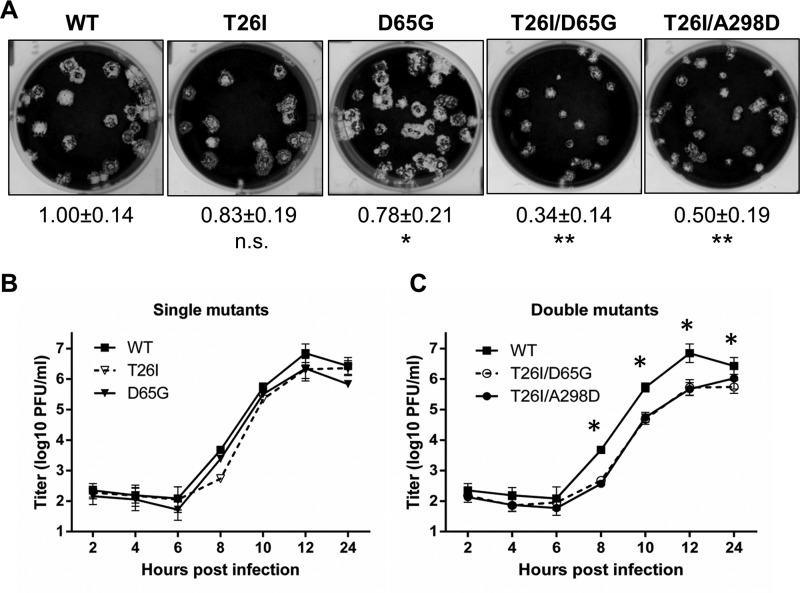
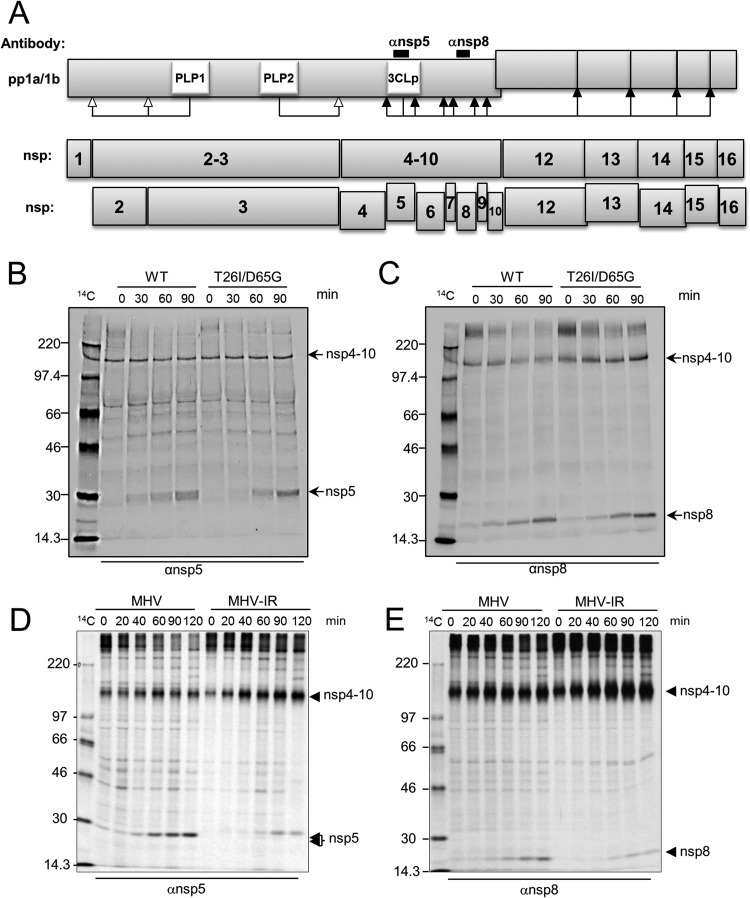
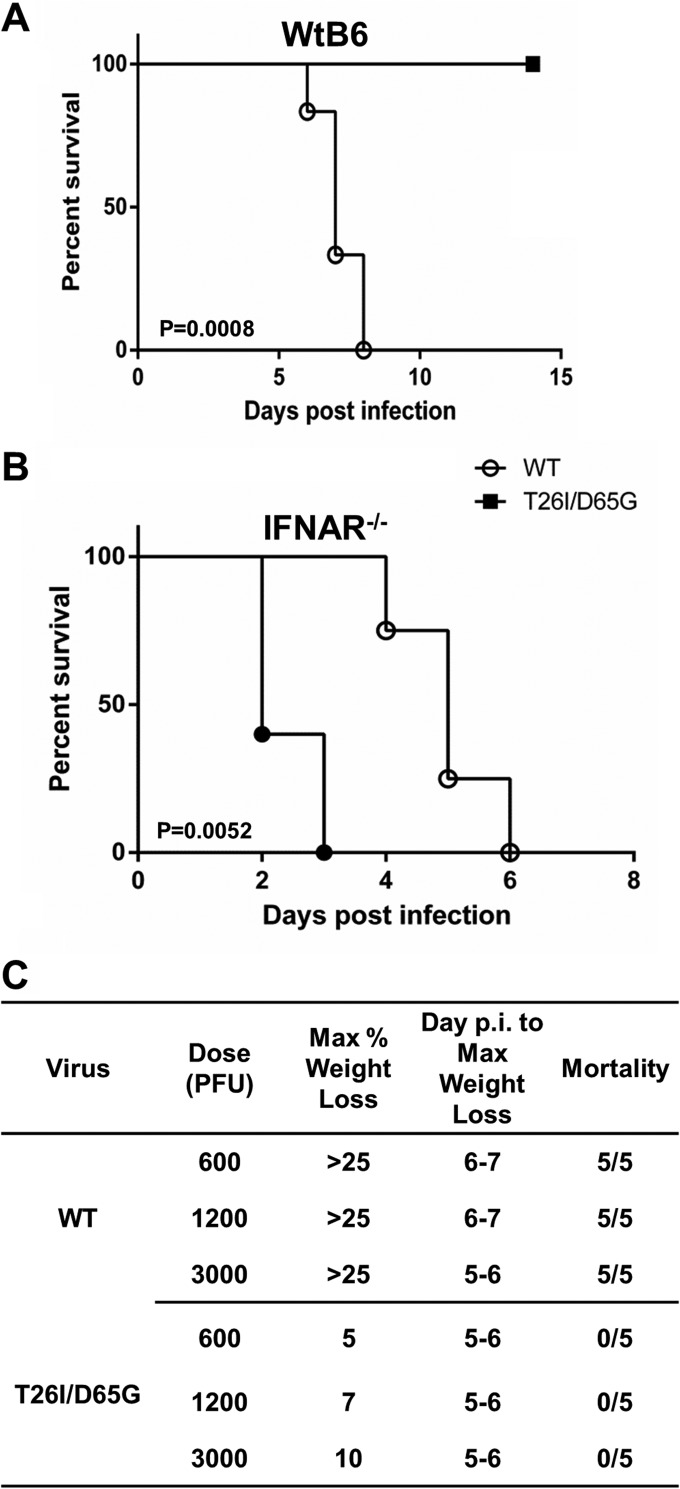
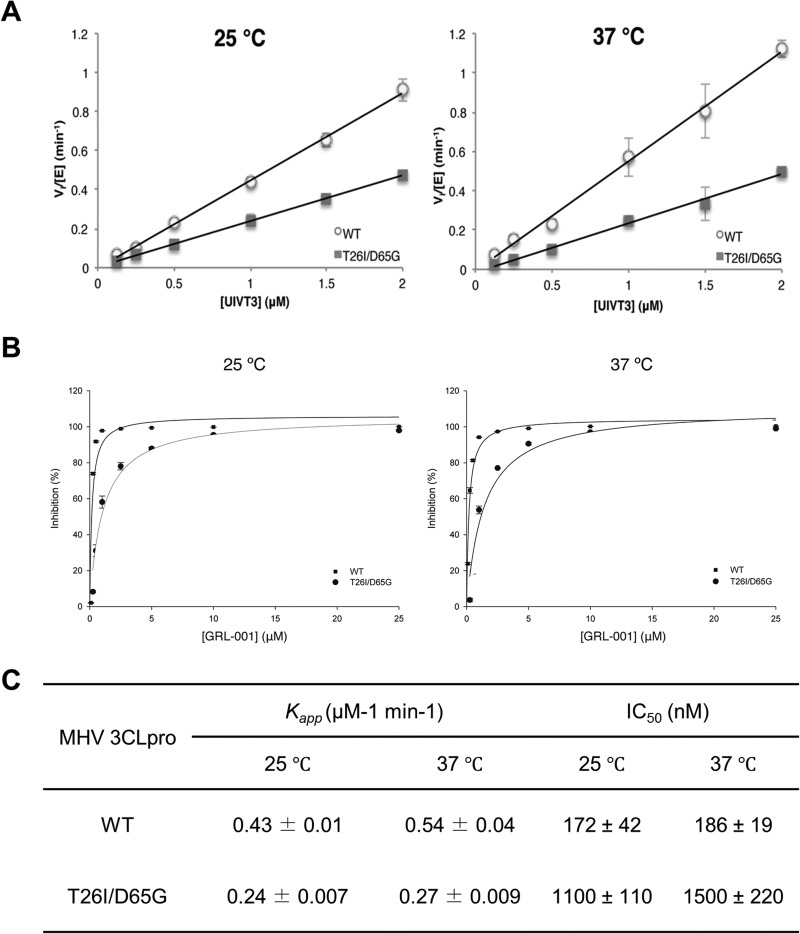
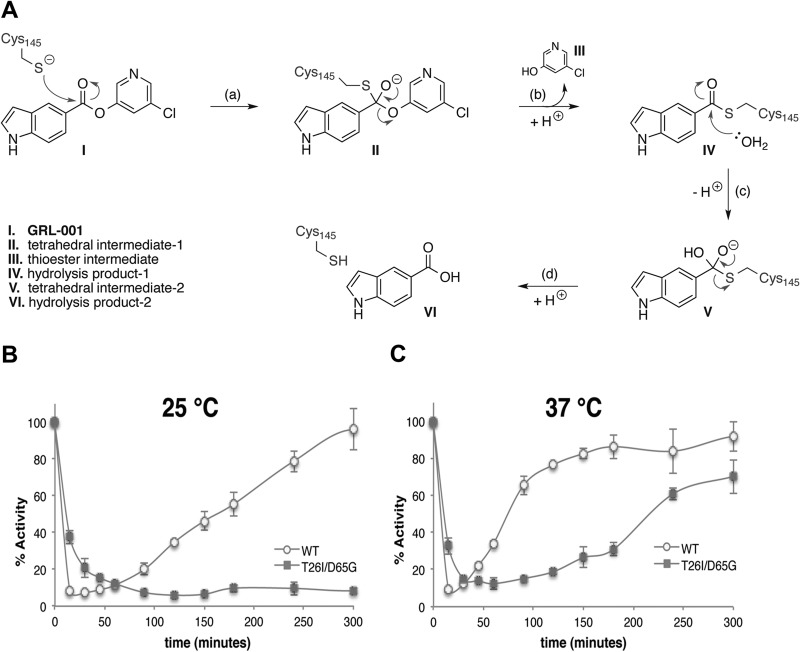
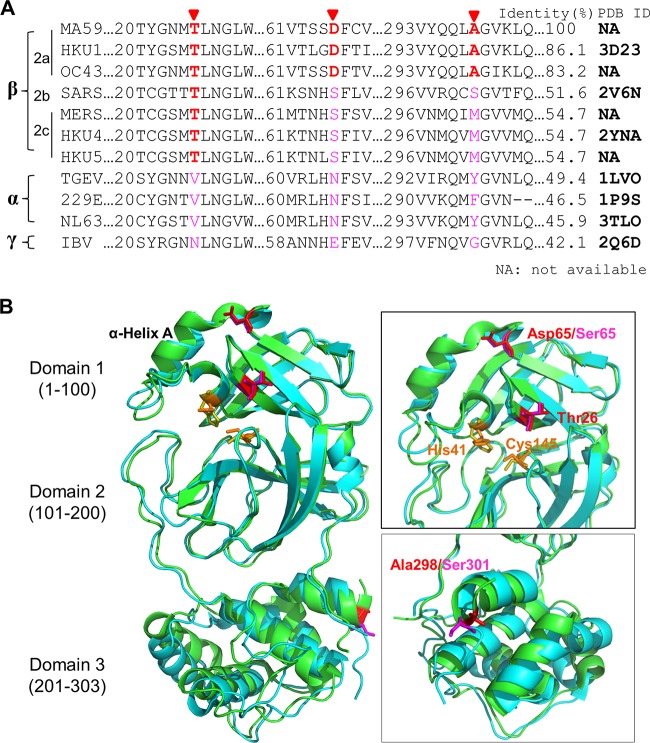
Viral protease inhibitors are remarkably effective at blocking the replication of viruses such as human immunodeficiency virus and hepatitis C virus, but they inevitably lead to the selection of inhibitor-resistant mutants, which may contribute to ongoing disease. Protease inhibitors blocking the replication of coronavirus (CoV), including the causative agents of severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS), provide a promising foundation for the development of anticoronaviral therapeutics. However, the selection and consequences of inhibitor-resistant CoVs are unknown. In this study, we exploited the model coronavirus, mouse hepatitis virus (MHV), to investigate the genotype and phenotype of MHV quasispecies selected for resistance to a broad-spectrum CoV 3C-like protease (3CLpro) inhibitor. Clonal sequencing identified single or double mutations within the 3CLpro coding sequence of inhibitor-resistant virus. Using reverse genetics to generate isogenic viruses with mutant 3CLpros, we found that viruses encoding double-mutant 3CLpros are fully resistant to the inhibitor and exhibit a significant delay in proteolytic processing of the viral replicase polyprotein. The inhibitor-resistant viruses also exhibited postponed and reduced production of infectious virus particles. Biochemical analysis verified double-mutant 3CLpro enzyme as impaired for protease activity and exhibiting reduced sensitivity to the inhibitor and revealed a delayed kinetics of inhibitor hydrolysis and activity restoration. Furthermore, the inhibitor-resistant virus was shown to be highly attenuated in mice. Our study provides the first insight into the pathogenicity and mechanism of 3CLpro inhibitor-resistant CoV mutants, revealing a low genetic barrier but high fitness cost of resistance. Importance: RNA viruses are infamous for their ability to evolve in response to selective pressure, such as the presence of antiviral drugs. For coronaviruses such as the causative agent of Middle East respiratory syndrome (MERS), protease inhibitors have been developed and shown to block virus replication, but the consequences of selection of inhibitor-resistant mutants have not been studied. Here, we report the low genetic barrier and relatively high deleterious consequences of CoV resistance to a 3CLpro protease inhibitor in a coronavirus model system, mouse hepatitis virus (MHV). We found that although mutations that confer resistance arise quickly, the resistant viruses replicate slowly and do not cause lethal disease in mice. Overall, our study provides the first analysis of the low barrier but high cost of resistance to a CoV 3CLpro inhibitor, which will facilitate the further development of protease inhibitors as anti-coronavirus therapeutics.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
Figures
References
-
- Hai R, Schmolke M, Leyva-Grado VH, Thangavel RR, Margine I, Jaffe EL, Krammer F, Solórzano A, García-Sastre A, Palese P, Bouvier NM. 2013. Influenza A(H7N9) virus gains neuraminidase inhibitor resistance without loss of in vivo virulence or transmissibility. Nat. Commun. 4:2854. 10.1038/ncomms3854 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
