

Next generation sequencing reveals skewing of the T and B cell receptor repertoires in patients with wiskott-Aldrich syndrome
- PMID: 25101082
- PMCID: PMC4102881
- DOI: 10.3389/fimmu.2014.00340
Next generation sequencing reveals skewing of the T and B cell receptor repertoires in patients with wiskott-Aldrich syndrome
Abstract
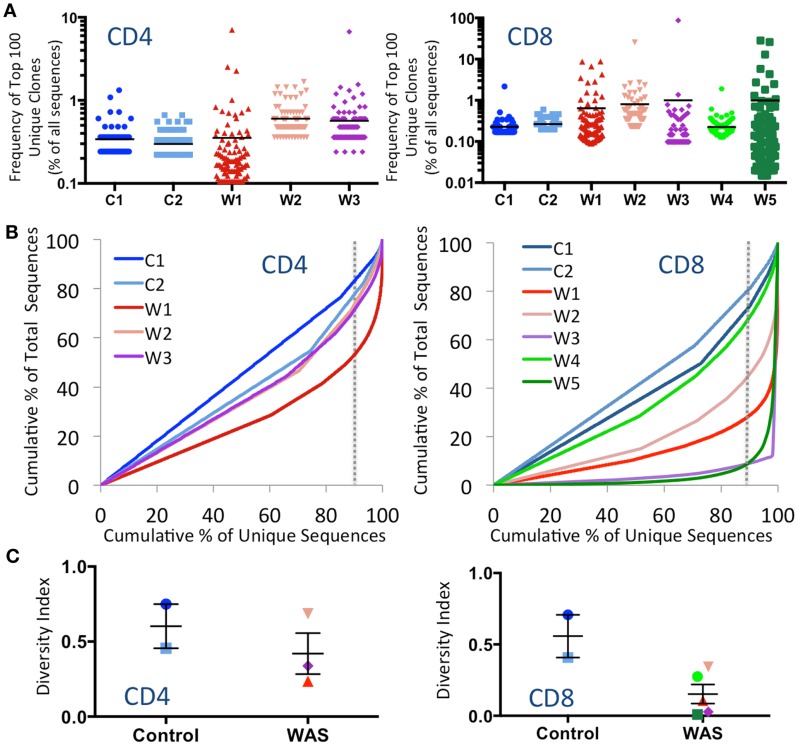
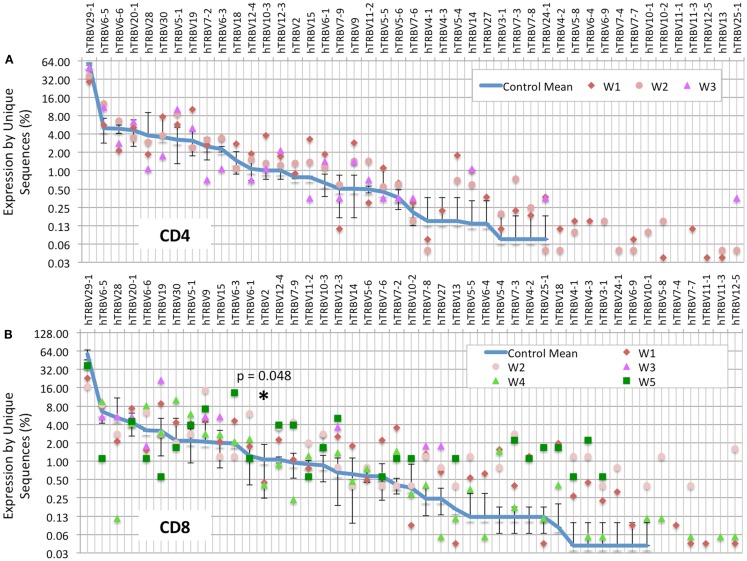
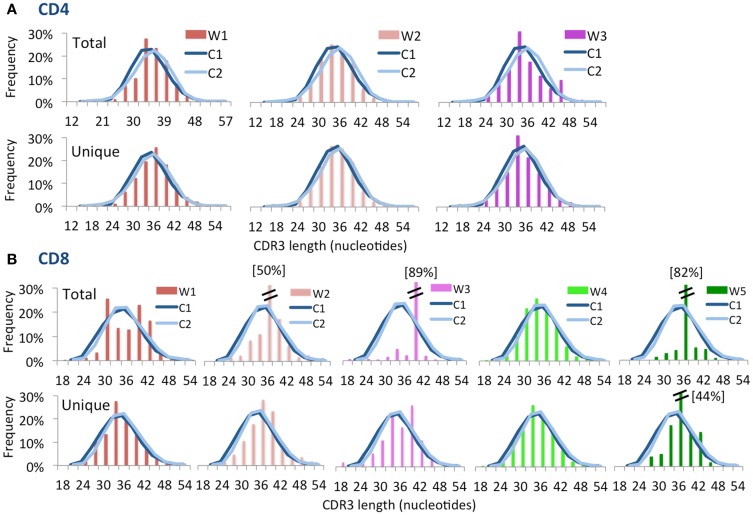
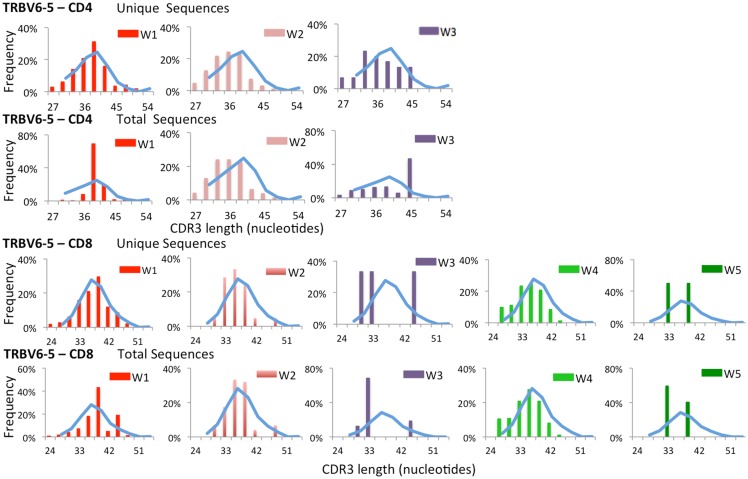
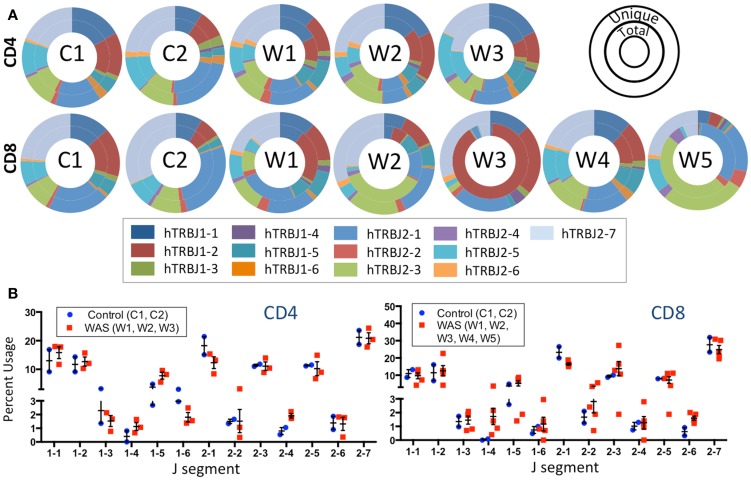
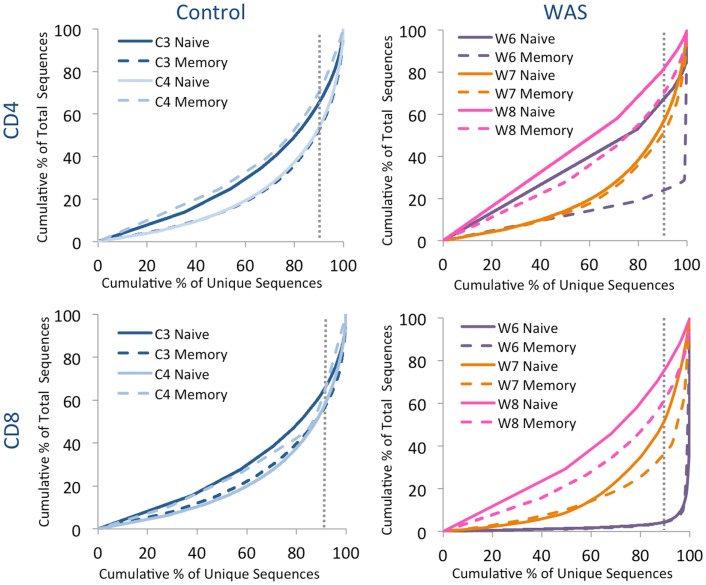
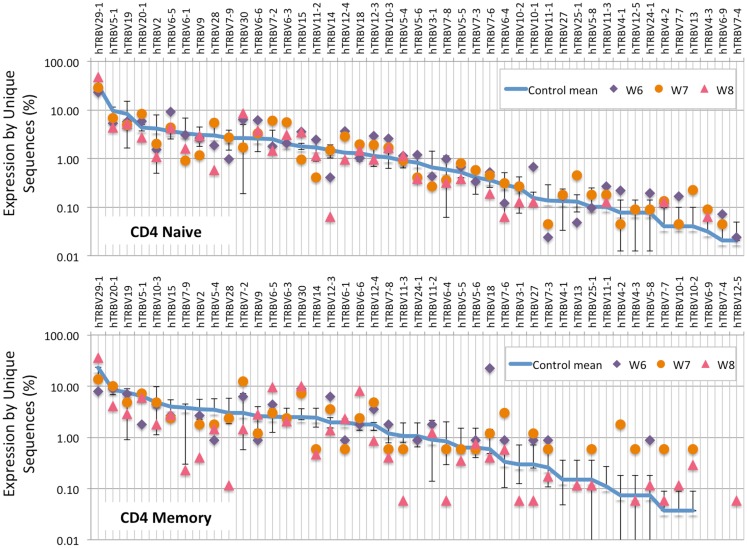
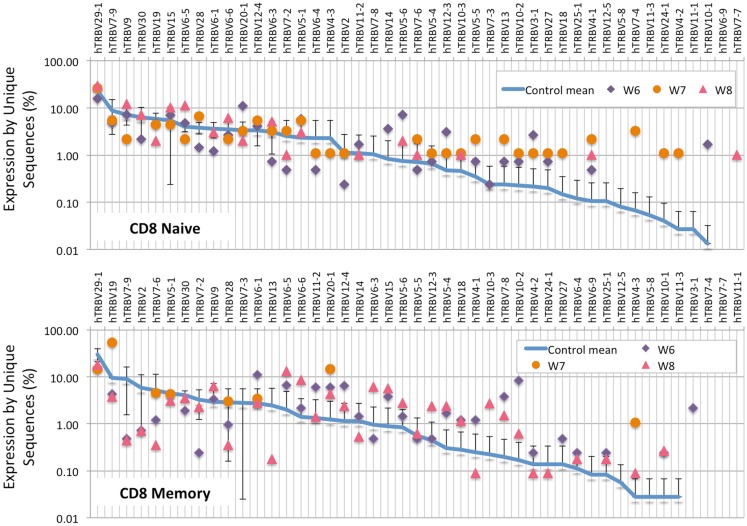
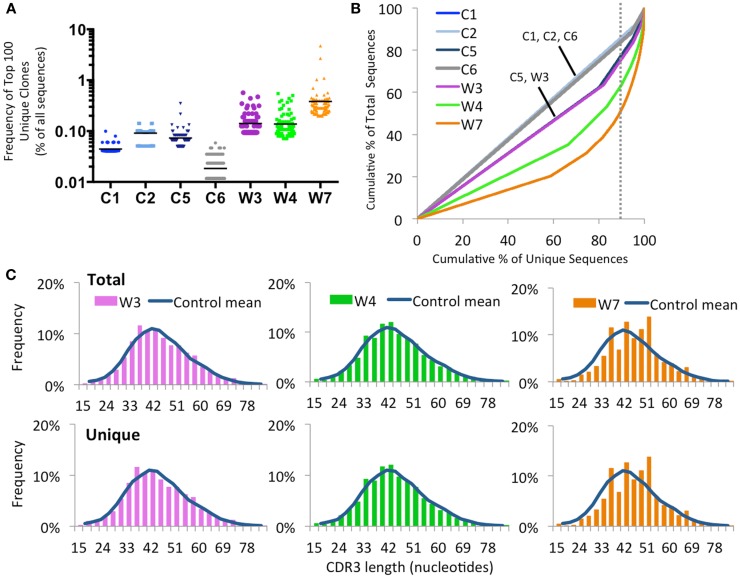
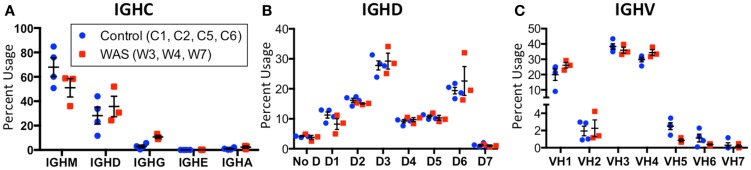
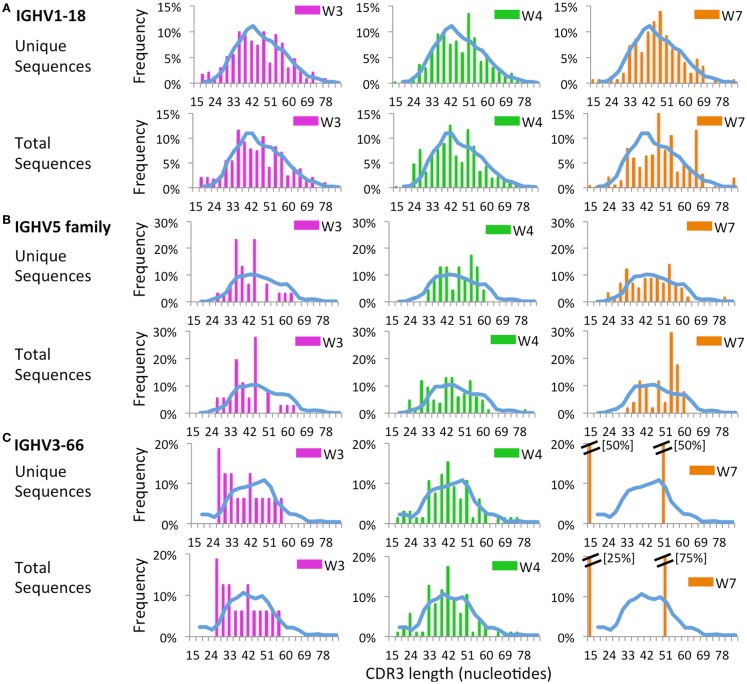
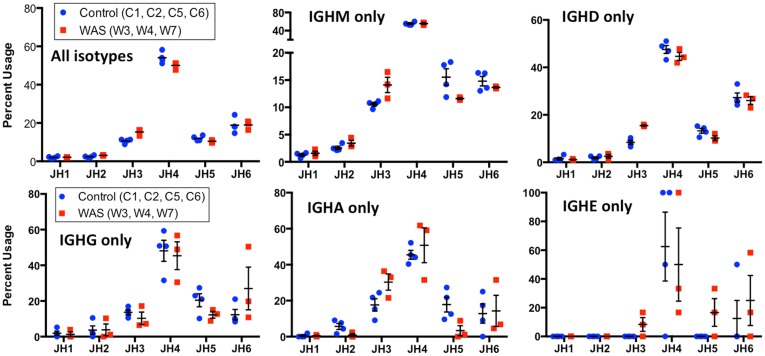
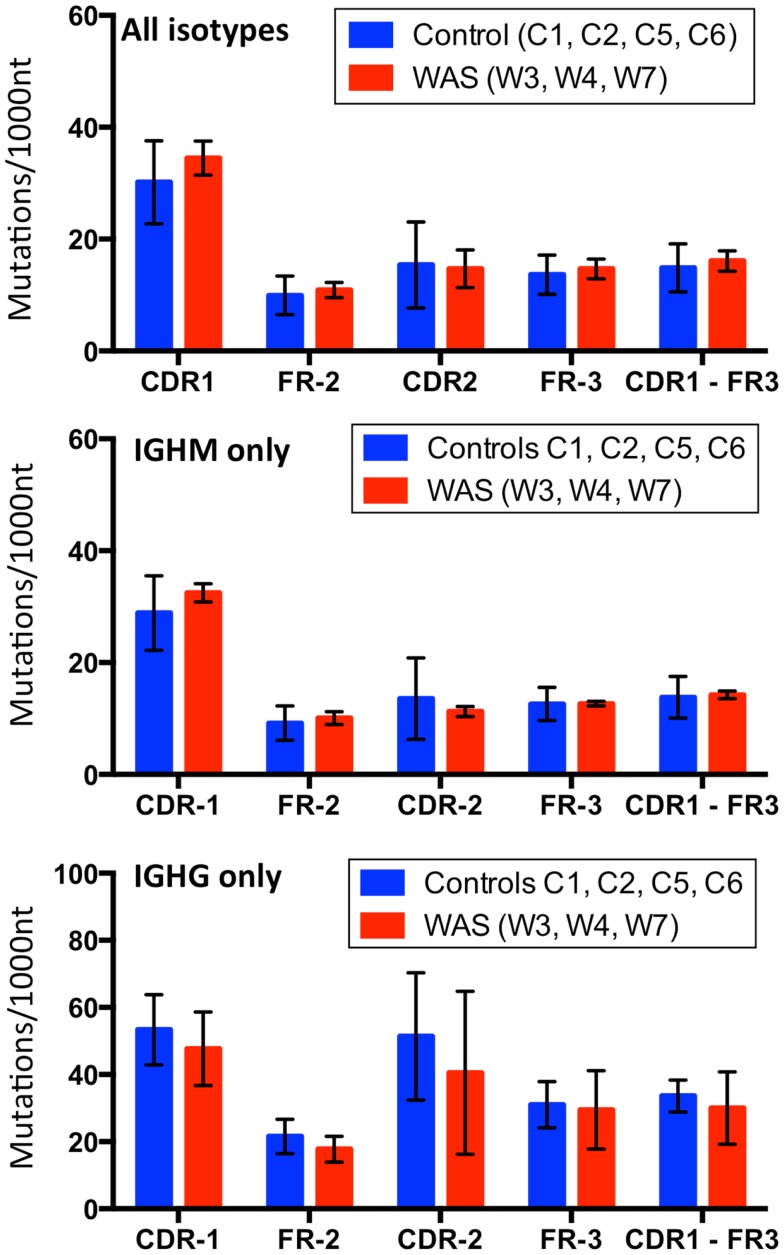
The Wiskott-Aldrich syndrome (WAS) is due to mutations of the WAS gene encoding for the cytoskeletal WAS protein, leading to abnormal downstream signaling from the T cell and B cell antigen receptors (TCR and BCR). We hypothesized that the impaired signaling through the TCR and BCR in WAS would subsequently lead to aberrations in the immune repertoire of WAS patients. Using next generation sequencing (NGS), the T cell receptor β and B cell immunoglobulin heavy chain (IGH) repertoires of eight patients with WAS and six controls were sequenced. Clonal expansions were identified within memory CD4(+) cells as well as in total, naïve and memory CD8(+) cells from WAS patients. In the B cell compartment, WAS patient IGH repertoires were also clonally expanded and showed skewed usage of IGHV and IGHJ genes, and increased usage of IGHG constant genes, compared with controls. To our knowledge, this is the first study that demonstrates significant abnormalities of the immune repertoire in WAS patients using NGS.
Keywords: B cell receptor; T cell receptor; Wiskott–Aldrich syndrome; clonotypic expansion; deep sequencing; immune repertoire; next generation sequencing; somatic hypermutation.
Figures

References
-
- Wiskott A. Familiarer, angeborener Morbus Werlhofii? Monatsschr Kinderheilkd (1937) 68:212–6
-
- Aldrich RA, Steinberg AG, Campbell DC. Pedigree demonstrating a sex-linked recessive condition characterized by draining ears, eczematoid dermatitis and bloody diarrhea. Pediatrics (1954) 13:133–9 - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials