

Endothelial C-type natriuretic peptide maintains vascular homeostasis
- PMID: 25105365
- PMCID: PMC4151218
- DOI: 10.1172/JCI74281
Endothelial C-type natriuretic peptide maintains vascular homeostasis
Abstract
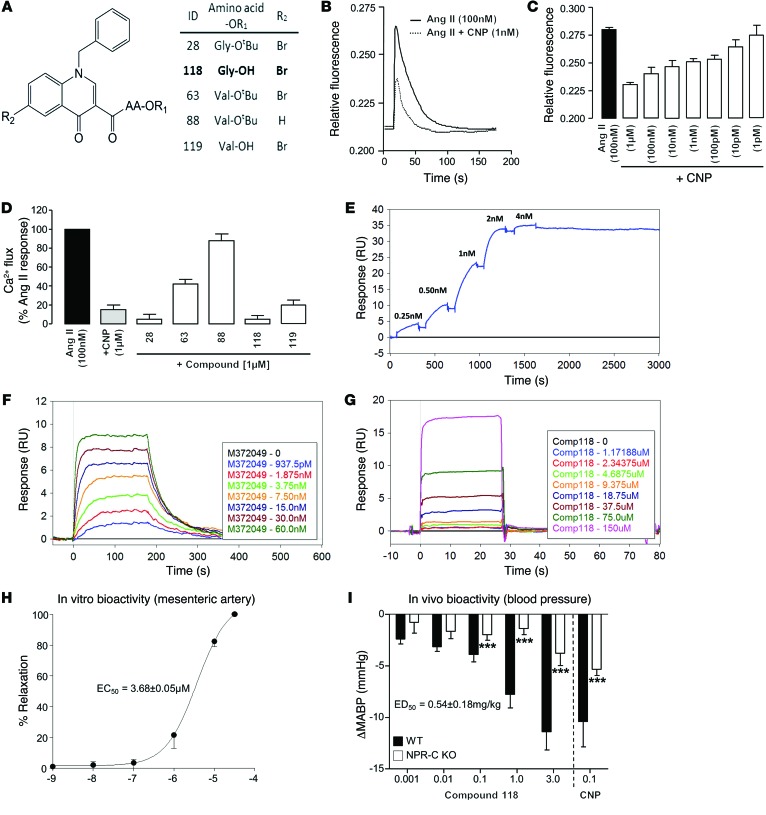
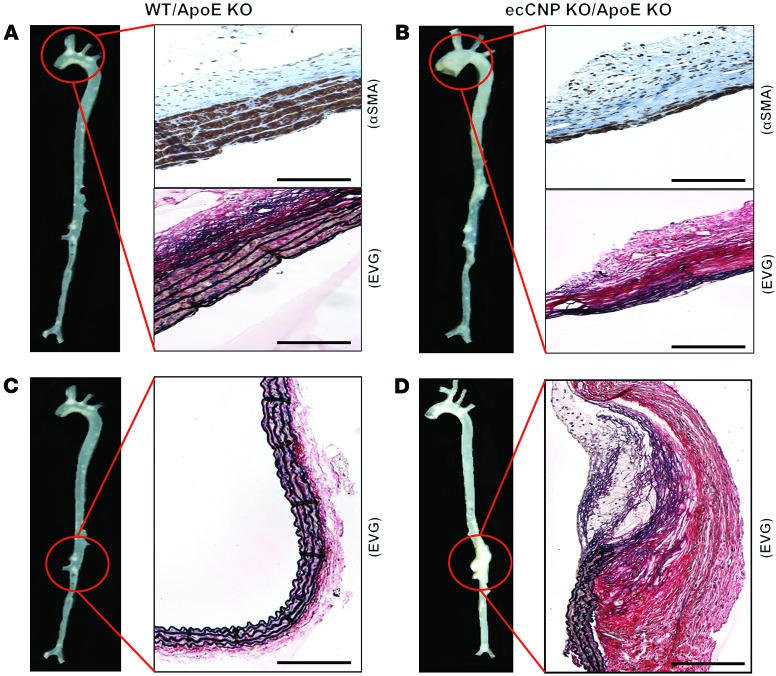
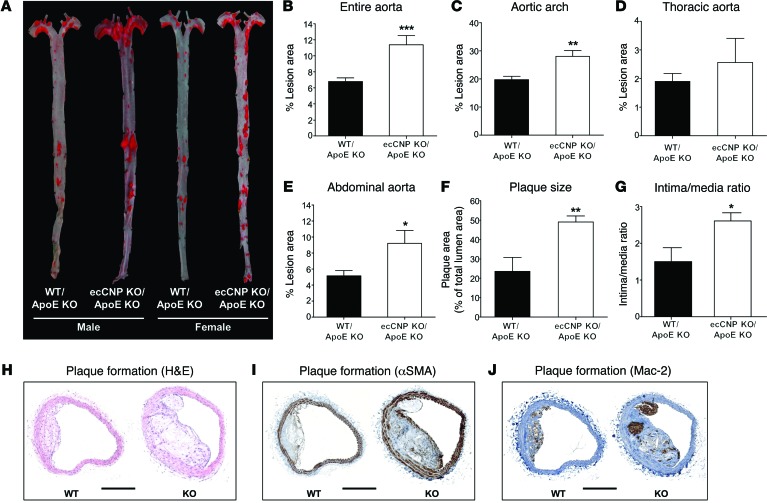
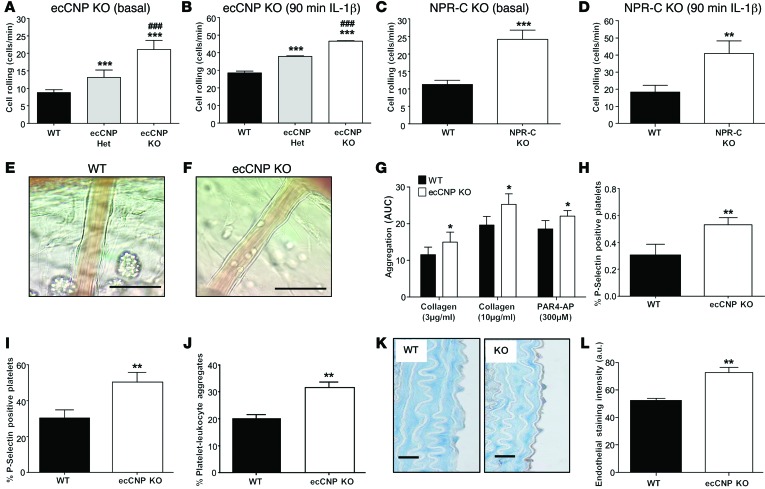
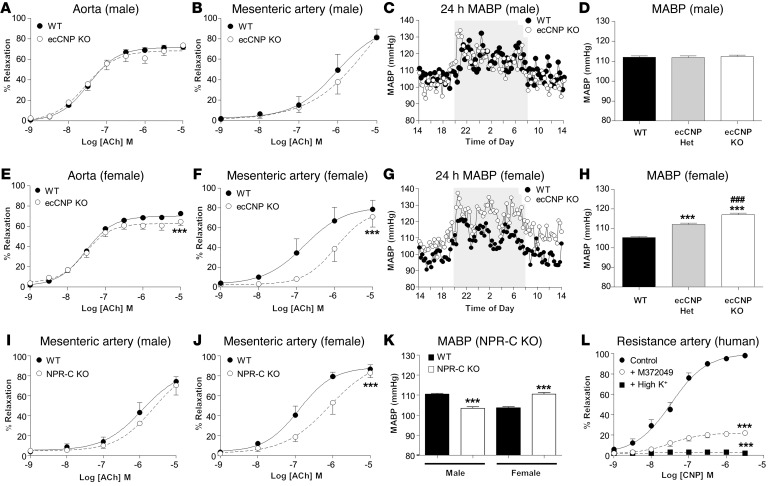
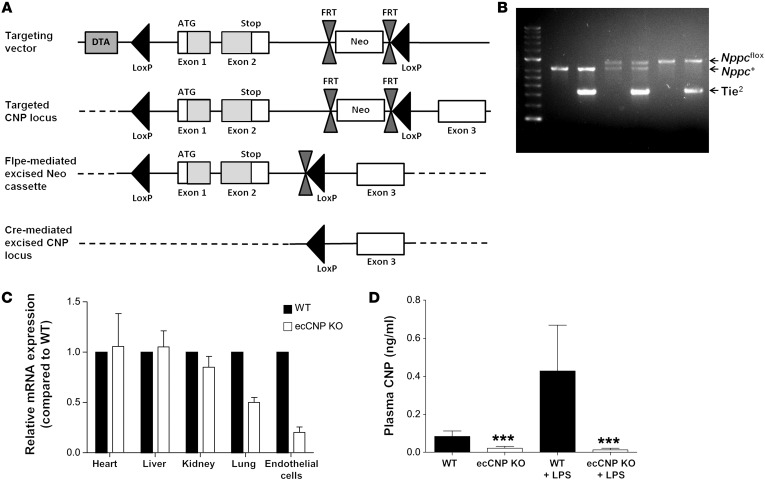
The endothelium plays a fundamental role in maintaining vascular homeostasis by releasing factors that regulate local blood flow, systemic blood pressure, and the reactivity of leukocytes and platelets. Accordingly, endothelial dysfunction underpins many cardiovascular diseases, including hypertension, myocardial infarction, and stroke. Herein, we evaluated mice with endothelial-specific deletion of Nppc, which encodes C-type natriuretic peptide (CNP), and determined that this mediator is essential for multiple aspects of vascular regulation. Specifically, disruption of CNP leads to endothelial dysfunction, hypertension, atherogenesis, and aneurysm. Moreover, we identified natriuretic peptide receptor-C (NPR-C) as the cognate receptor that primarily underlies CNP-dependent vasoprotective functions and developed small-molecule NPR-C agonists to target this pathway. Administration of NPR-C agonists promotes a vasorelaxation of isolated resistance arteries and a reduction in blood pressure in wild-type animals that is diminished in mice lacking NPR-C. This work provides a mechanistic explanation for genome-wide association studies that have linked the NPR-C (Npr3) locus with hypertension by demonstrating the importance of CNP/NPR-C signaling in preserving vascular homoeostasis. Furthermore, these results suggest that the CNP/NPR-C pathway has potential as a disease-modifying therapeutic target for cardiovascular disorders.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
