

Lean Big Data integration in systems biology and systems pharmacology
- PMID: 25109570
- PMCID: PMC4153537
- DOI: 10.1016/j.tips.2014.07.001
Lean Big Data integration in systems biology and systems pharmacology
Abstract
Data sets from recent large-scale projects can be integrated into one unified puzzle that can provide new insights into how drugs and genetic perturbations applied to human cells are linked to whole-organism phenotypes. Data that report how drugs affect the phenotype of human cell lines and how drugs induce changes in gene and protein expression in human cell lines can be combined with knowledge about human disease, side effects induced by drugs, and mouse phenotypes. Such data integration efforts can be achieved through the conversion of data from the various resources into single-node-type networks, gene-set libraries, or multipartite graphs. This approach can lead us to the identification of more relationships between genes, drugs, and phenotypes as well as benchmark computational and experimental methods. Overall, this lean 'Big Data' integration strategy will bring us closer toward the goal of realizing personalized medicine.
Keywords: data integration; network analysis; network pharmacology; side-effect prediction; systems pharmacology; target prediction.
Copyright © 2014 Elsevier Ltd. All rights reserved.
Figures






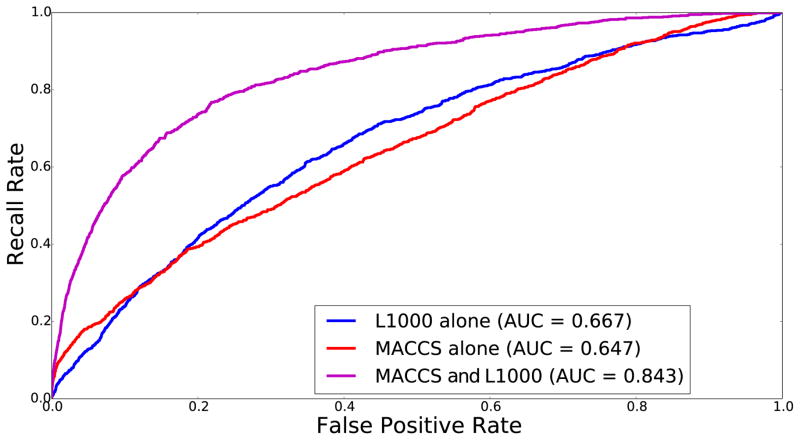
References
-
- Mayer-Schönberger V, Cukier K. Big data: A revolution that will transform how we live, work, and think. Houghton Mifflin Harcourt; 2013.
-
- Chibon F. Cancer gene expression signatures–The rise and fall? European Journal of Cancer. 2013;49:2000–2009. - PubMed
-
- Stegmaier K, Ross KN, Colavito SA, O’Malley S, Stockwell BR, Golub TR. Gene expression–based high-throughput screening (GE-HTS) and application to leukemia differentiation. Nature genetics. 2004;36:257–263. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
