

The first knockin mouse model of episodic ataxia type 2
- PMID: 25109669
- PMCID: PMC4194266
- DOI: 10.1016/j.expneurol.2014.08.001
The first knockin mouse model of episodic ataxia type 2
Abstract
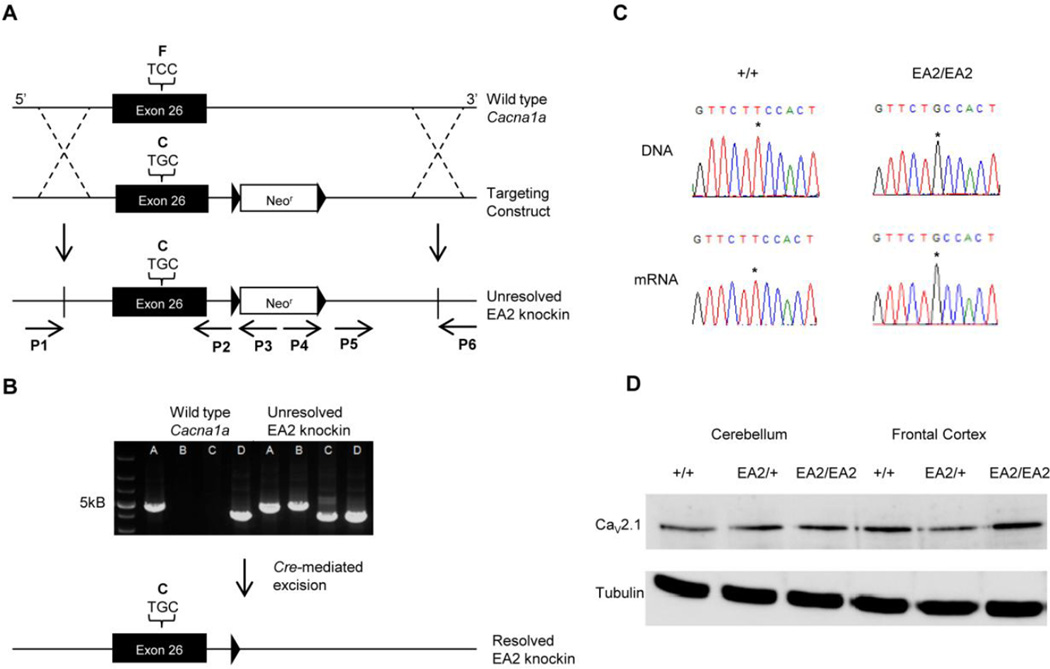
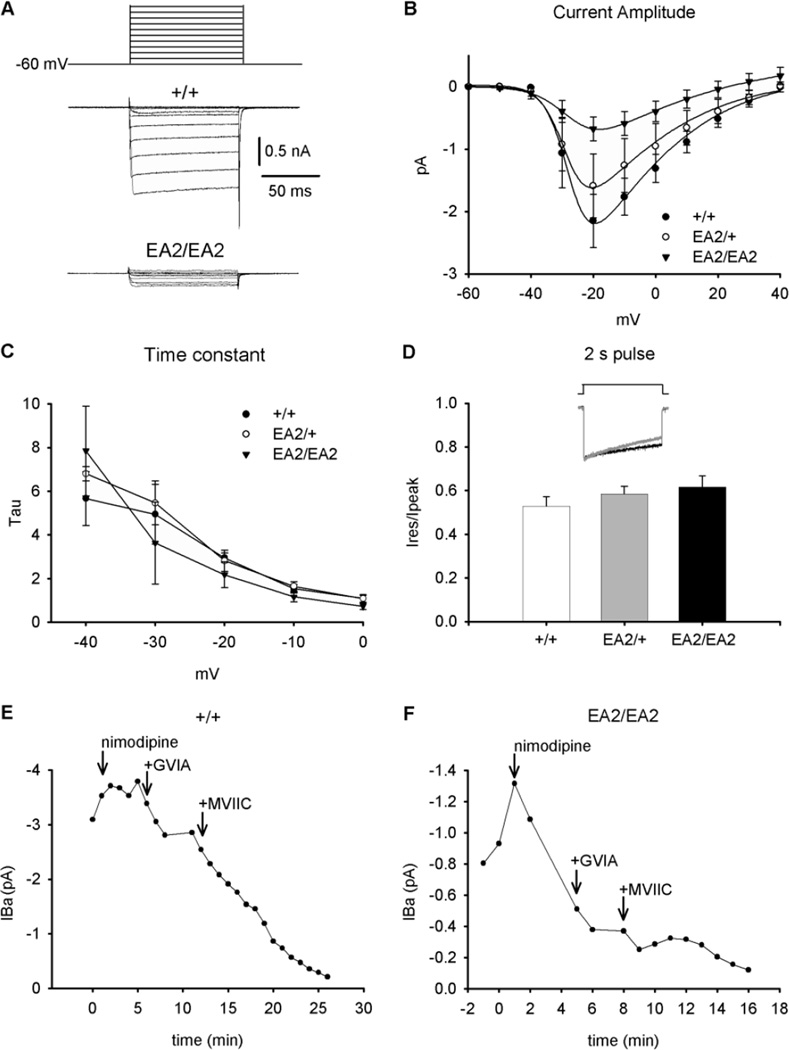
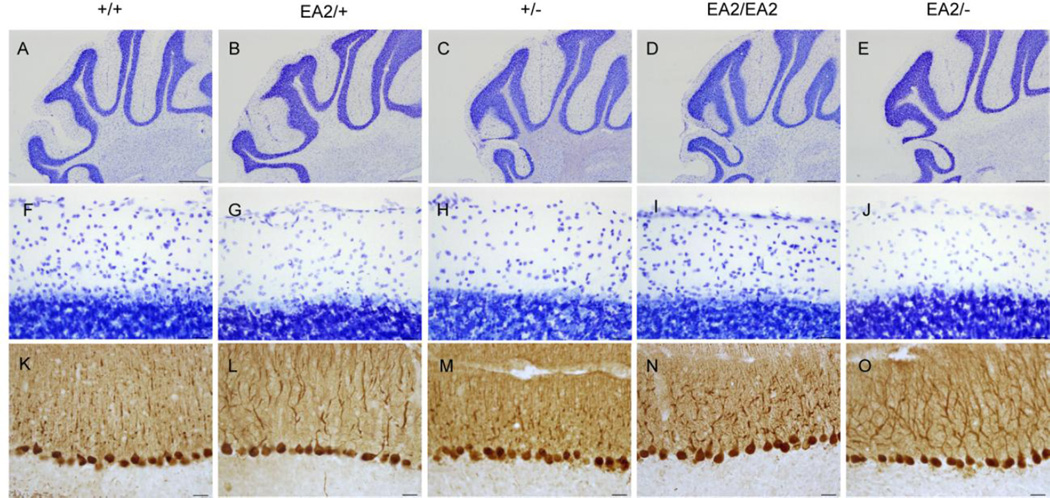
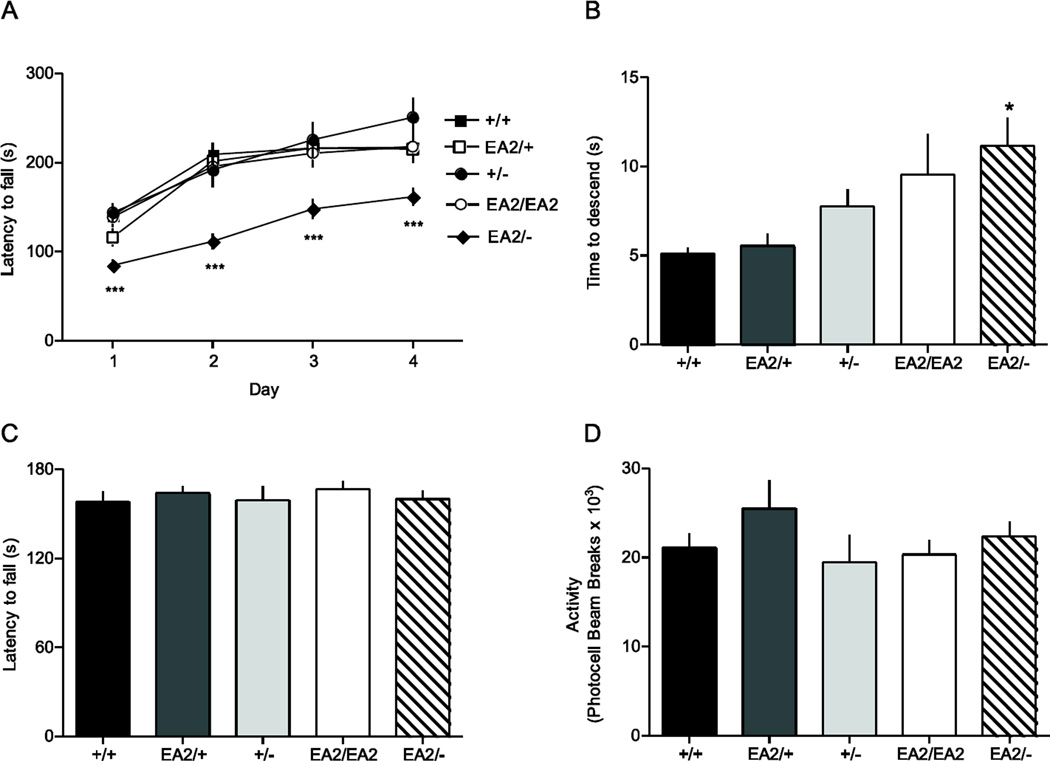
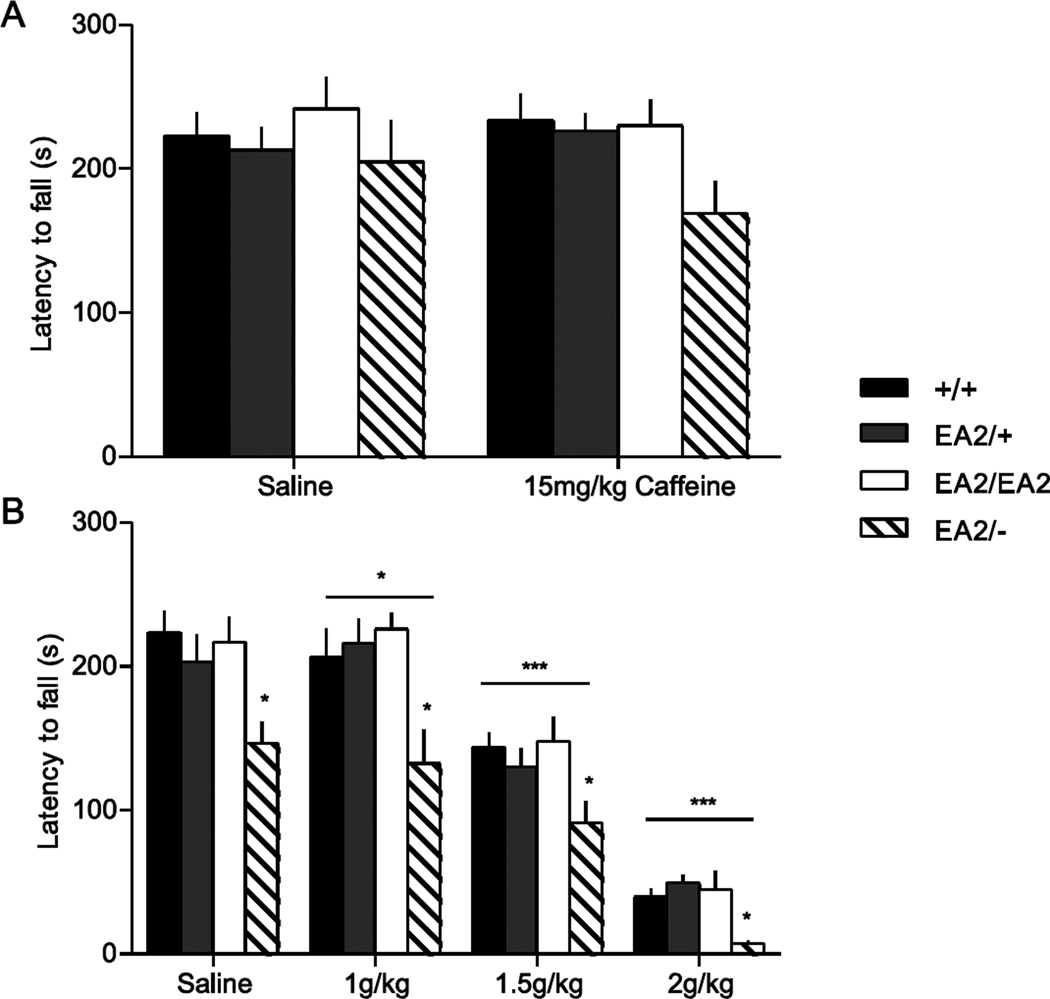
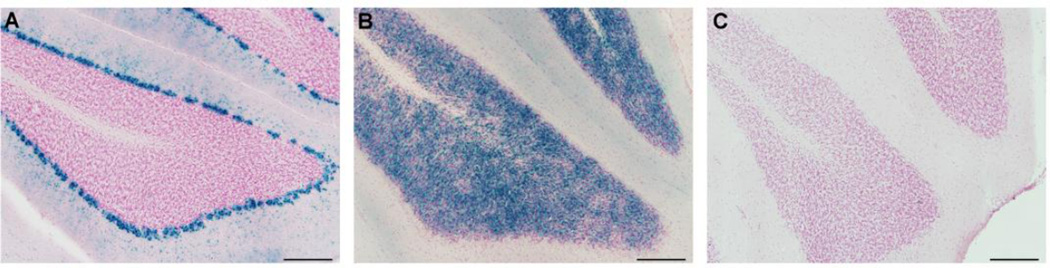
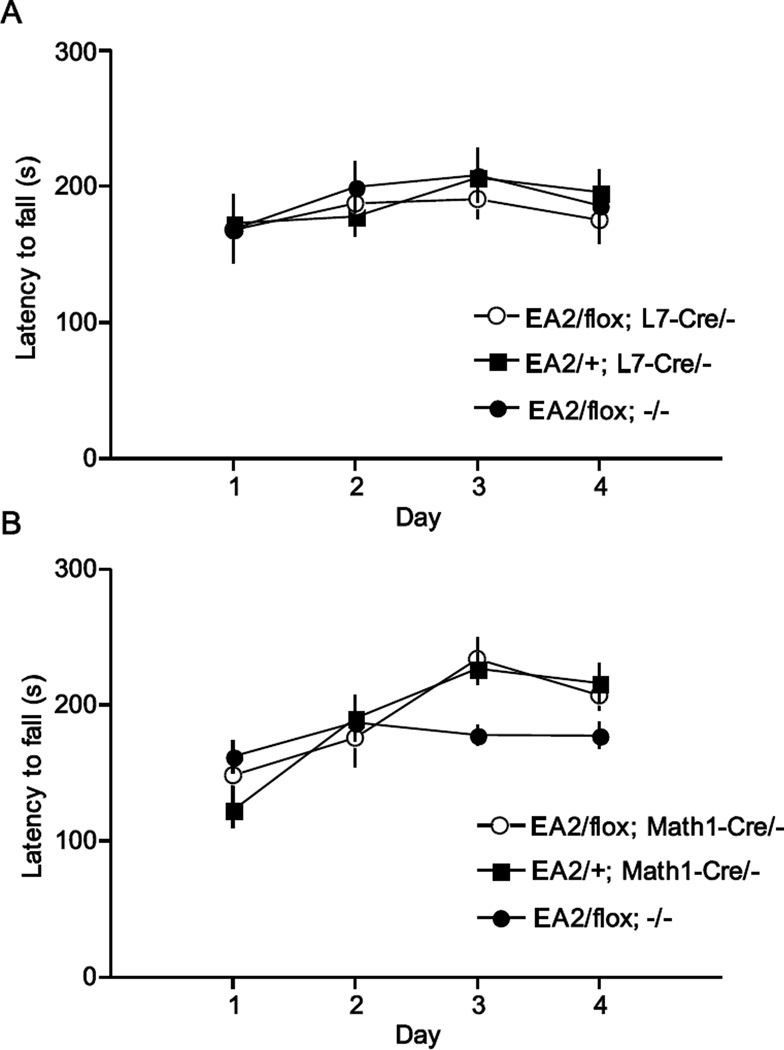
Episodic ataxia type 2 (EA2) is an autosomal dominant disorder associated with attacks of ataxia that are typically precipitated by stress, ethanol, caffeine or exercise. EA2 is caused by loss-of-function mutations in the CACNA1A gene, which encodes the α1A subunit of the CaV2.1 voltage-gated Ca(2+) channel. To better understand the pathomechanisms of this disorder in vivo, we created the first genetic animal model of EA2 by engineering a mouse line carrying the EA2-causing c.4486T>G (p.F1406C) missense mutation in the orthologous mouse Cacna1a gene. Mice homozygous for the mutated allele exhibit a ~70% reduction in CaV2.1 current density in Purkinje cells, though surprisingly do not exhibit an overt motor phenotype. Mice hemizygous for the knockin allele (EA2/- mice) did exhibit motor dysfunction measurable by rotarod and pole test. Studies using Cre-flox conditional genetics explored the role of cerebellar Purkinje cells or cerebellar granule cells in the poor motor performance of EA2/- mice and demonstrate that manipulation of either cell type alone did not cause poor motor performance. Thus, it is possible that subtle dysfunction arising from multiple cell types is necessary for the expression of certain ataxia syndromes.
Keywords: Ataxia; CACNA1A; Cerebellar granule cell; Channelopathy; Cre; EA2; Episodic ataxia type 2; Knockin; Purkinje cell; Voltage-gated calcium channel.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Ubiquitin Ligase RNF138 Promotes Episodic Ataxia Type 2-Associated Aberrant Degradation of Human Cav2.1 (P/Q-Type) Calcium Channels.J Neurosci. 2017 Mar 1;37(9):2485-2503. doi: 10.1523/JNEUROSCI.3070-16.2017. Epub 2017 Feb 6. J Neurosci. 2017. PMID: 28167673 Free PMC article.
-
RNAi silencing of P/Q-type calcium channels in Purkinje neurons of adult mouse leads to episodic ataxia type 2.Neurobiol Dis. 2014 Aug;68:47-56. doi: 10.1016/j.nbd.2014.04.005. Epub 2014 Apr 21. Neurobiol Dis. 2014. PMID: 24768804
-
Characterization of the dominant inheritance mechanism of Episodic Ataxia type 2.Neurobiol Dis. 2017 Oct;106:110-123. doi: 10.1016/j.nbd.2017.07.004. Epub 2017 Jul 5. Neurobiol Dis. 2017. PMID: 28688851
-
Epilepsy and episodic ataxia type 2: family study and review of the literature.J Neurol. 2021 Nov;268(11):4296-4302. doi: 10.1007/s00415-021-10555-0. Epub 2021 May 13. J Neurol. 2021. PMID: 33983550 Review.
-
Episodic ataxias 1 and 2.Handb Clin Neurol. 2012;103:595-602. doi: 10.1016/B978-0-444-51892-7.00042-5. Handb Clin Neurol. 2012. PMID: 21827920 Review.
Cited by
-
CACNA1A haploinsufficiency leads to reduced synaptic function and increased intrinsic excitability.Brain. 2025 Apr 3;148(4):1286-1301. doi: 10.1093/brain/awae330. Brain. 2025. PMID: 39460936 Free PMC article.
-
What Is the Role of Norepinephrine in Cerebellar Modulation and Stress-Induced Episodic Ataxia?Neurology. 2023 Feb 21;100(8):383-386. doi: 10.1212/WNL.0000000000206882. Neurology. 2023. PMID: 36806456 Free PMC article. No abstract available.
-
Mitochondrial Dysfunction may explain symptom variation in Phelan-McDermid Syndrome.Sci Rep. 2016 Jan 29;6:19544. doi: 10.1038/srep19544. Sci Rep. 2016. PMID: 26822410 Free PMC article.
-
Overlaps, gaps, and complexities of mouse models of Developmental and Epileptic Encephalopathy.Neurobiol Dis. 2021 Jan;148:105220. doi: 10.1016/j.nbd.2020.105220. Epub 2020 Dec 7. Neurobiol Dis. 2021. PMID: 33301879 Free PMC article. Review.
-
Ubiquitin Ligase RNF138 Promotes Episodic Ataxia Type 2-Associated Aberrant Degradation of Human Cav2.1 (P/Q-Type) Calcium Channels.J Neurosci. 2017 Mar 1;37(9):2485-2503. doi: 10.1523/JNEUROSCI.3070-16.2017. Epub 2017 Feb 6. J Neurosci. 2017. PMID: 28167673 Free PMC article.
References
-
- Baloh RW. Episodic ataxias 1 and 2. Handb Clin Neurol. 2012;103:595–602. - PubMed
-
- Barski JJ, Dethleffsen K, Meyer M. Cre recombinase expression in cerebellar Purkinje cells. Genesis. 2000;28:93–98. - PubMed
-
- Bhatia KP, Griggs RC, Ptacek LJ. Episodic movement disorders as channelopathies. Mov Disord. 2000;15:429–433. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
