

DNA methylation modifications associated with chronic fatigue syndrome
- PMID: 25111603
- PMCID: PMC4128721
- DOI: 10.1371/journal.pone.0104757
DNA methylation modifications associated with chronic fatigue syndrome
Abstract
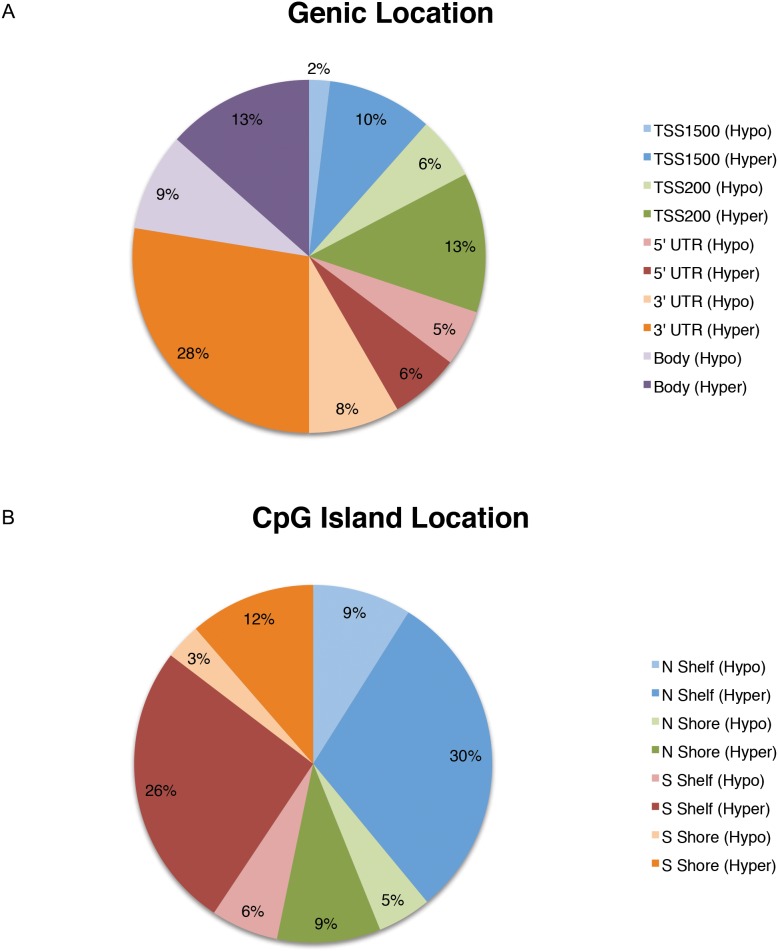
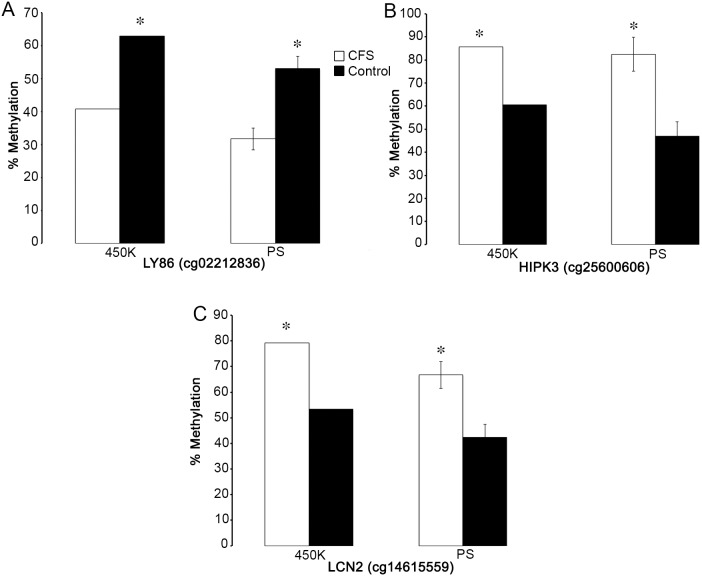
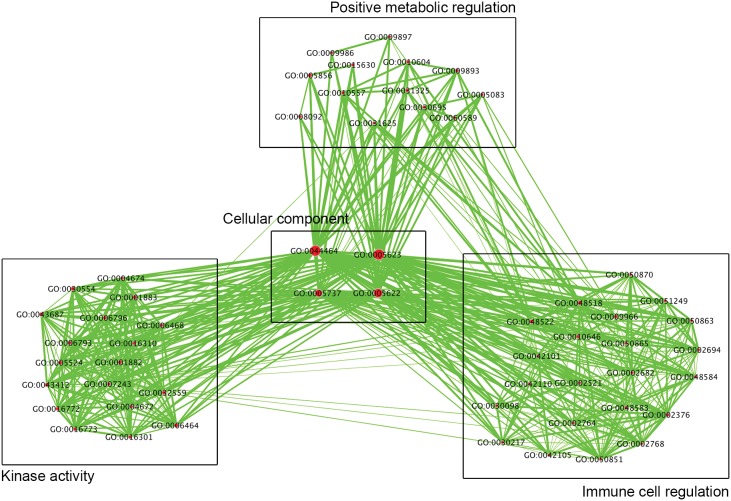
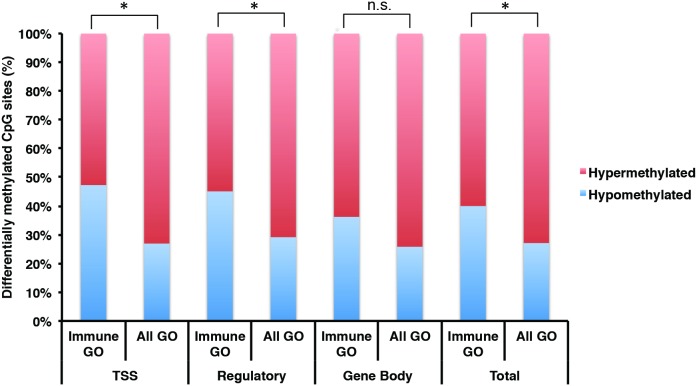
Chronic Fatigue Syndrome (CFS), also known as myalgic encephalomyelitis, is a complex multifactorial disease that is characterized by the persistent presence of fatigue and other particular symptoms for a minimum of 6 months. Symptoms fail to dissipate after sufficient rest and have major effects on the daily functioning of CFS sufferers. CFS is a multi-system disease with a heterogeneous patient population showing a wide variety of functional disabilities and its biological basis remains poorly understood. Stable alterations in gene function in the immune system have been reported in several studies of CFS. Epigenetic modifications have been implicated in long-term effects on gene function, however, to our knowledge, genome-wide epigenetic modifications associated with CFS have not been explored. We examined the DNA methylome in peripheral blood mononuclear cells isolated from CFS patients and healthy controls using the Illumina HumanMethylation450 BeadChip array, controlling for invariant probes and probes overlapping polymorphic sequences. Gene ontology (GO) and network analysis of differentially methylated genes was performed to determine potential biological pathways showing changes in DNA methylation in CFS. We found an increased abundance of differentially methylated genes related to the immune response, cellular metabolism, and kinase activity. Genes associated with immune cell regulation, the largest coordinated enrichment of differentially methylated pathways, showed hypomethylation within promoters and other gene regulatory elements in CFS. These data are consistent with evidence of multisystem dysregulation in CFS and implicate the involvement of DNA modifications in CFS pathology.
Conflict of interest statement
Figures
References
-
- Fukuda K, Straus SE, Hickie I, Sharpe MC, Dobbins JG, et al. (1994) The chronic fatigue syndrome: a comprehensive approach to its definition and study. International Chronic Fatigue Syndrome Study Group. Ann Intern Med 121: 953–959. - PubMed
-
- Jason LA, Corradi K, Torres-Harding S, Taylor RR, King C (2005) Chronic fatigue syndrome: the need for subtypes. Neuropsychol Rev 15: 29–58. - PubMed
-
- Vernon SD, Reeves WC (2006) The challenge of integrating disparate high-content data: epidemiological, clinical and laboratory data collected during an in-hospital study of chronic fatigue syndrome. Pharmacogenomics 7: 345–354. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
