

Impact of pre-imputation SNP-filtering on genotype imputation results
- PMID: 25112433
- PMCID: PMC4236550
- DOI: 10.1186/s12863-014-0088-5
Impact of pre-imputation SNP-filtering on genotype imputation results
Abstract
Background: Imputation of partially missing or unobserved genotypes is an indispensable tool for SNP data analyses. However, research and understanding of the impact of initial SNP-data quality control on imputation results is still limited. In this paper, we aim to evaluate the effect of different strategies of pre-imputation quality filtering on the performance of the widely used imputation algorithms MaCH and IMPUTE.
Results: We considered three scenarios: imputation of partially missing genotypes with usage of an external reference panel, without usage of an external reference panel, as well as imputation of completely un-typed SNPs using an external reference panel. We first created various datasets applying different SNP quality filters and masking certain percentages of randomly selected high-quality SNPs. We imputed these SNPs and compared the results between the different filtering scenarios by using established and newly proposed measures of imputation quality. While the established measures assess certainty of imputation results, our newly proposed measures focus on the agreement with true genotypes. These measures showed that pre-imputation SNP-filtering might be detrimental regarding imputation quality. Moreover, the strongest drivers of imputation quality were in general the burden of missingness and the number of SNPs used for imputation. We also found that using a reference panel always improves imputation quality of partially missing genotypes. MaCH performed slightly better than IMPUTE2 in most of our scenarios. Again, these results were more pronounced when using our newly defined measures of imputation quality.
Conclusion: Even a moderate filtering has a detrimental effect on the imputation quality. Therefore little or no SNP filtering prior to imputation appears to be the best strategy for imputing small to moderately sized datasets. Our results also showed that for these datasets, MaCH performs slightly better than IMPUTE2 in most scenarios at the cost of increased computing time.
Figures
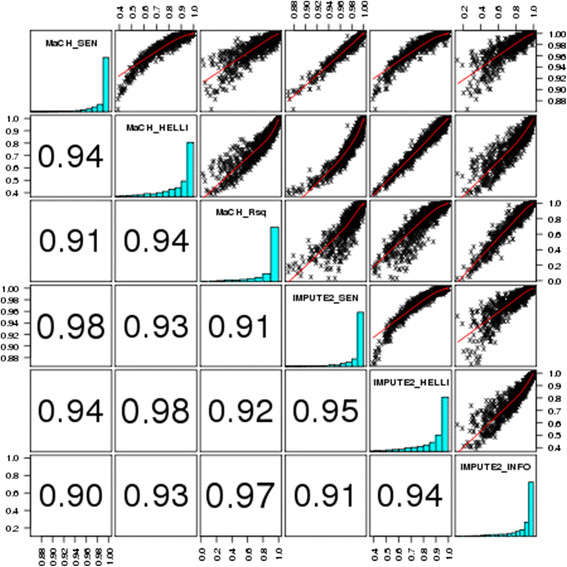
References
-
- Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, Leal SM, Pasternak S, Wheeler DA, Willis TD, Yu F, Yang H, Zeng C, Gao Y, Hu H, Hu W, Li C, Lin W, Liu S, Pan H, Tang X, Wang J, Wang W, Yu J, Zhang B, Zhang Q. et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
