

The roles of the RIIβ linker and N-terminal cyclic nucleotide-binding domain in determining the unique structures of the type IIβ protein kinase A: a small angle x-ray and neutron scattering study
- PMID: 25112875
- PMCID: PMC4192500
- DOI: 10.1074/jbc.M114.584177
The roles of the RIIβ linker and N-terminal cyclic nucleotide-binding domain in determining the unique structures of the type IIβ protein kinase A: a small angle x-ray and neutron scattering study
Abstract
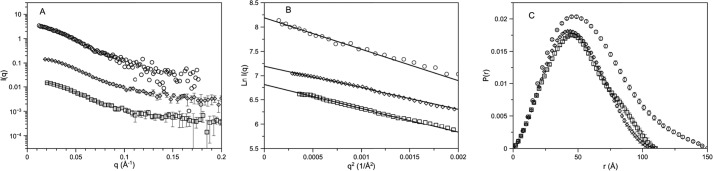
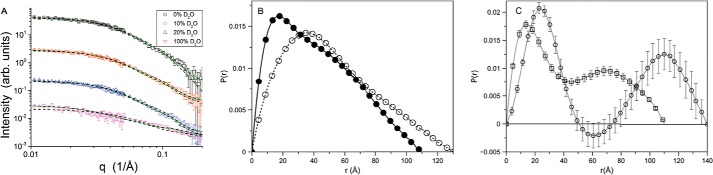
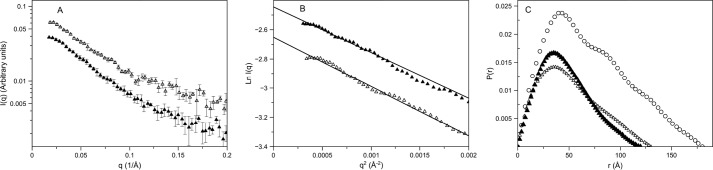
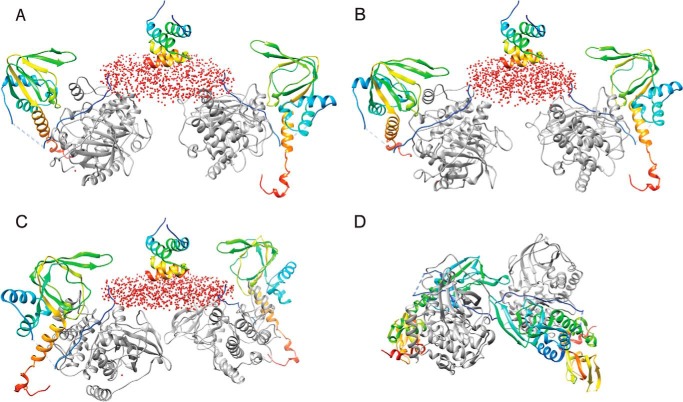
Protein kinase A (PKA) is ubiquitously expressed and is responsible for regulating many important cellular functions in response to changes in intracellular cAMP concentrations. The PKA holoenzyme is a tetramer (R2:C2), with a regulatory subunit homodimer (R2) that binds and inhibits two catalytic (C) subunits; binding of cAMP to the regulatory subunit homodimer causes activation of the catalytic subunits. Four different R subunit isoforms exist in mammalian cells, and these confer different structural features, subcellular localization, and biochemical properties upon the PKA holoenzymes they form. The holoenzyme containing RIIβ is structurally unique in that the type IIβ holoenzyme is much more compact than the free RIIβ homodimer. We have used small angle x-ray scattering and small angle neutron scattering to study the solution structure and subunit organization of a holoenzyme containing an RIIβ C-terminal deletion mutant (RIIβ(1-280)), which is missing the C-terminal cAMP-binding domain to better understand the structural organization of the type IIβ holoenzyme and the RIIβ domains that contribute to stabilizing the holoenzyme conformation. Our results demonstrate that compaction of the type IIβ holoenzyme does not require the C-terminal cAMP-binding domain but rather involves large structural rearrangements within the linker and N-terminal cyclic nucleotide-binding domain of the RIIβ homodimer. The structural rearrangements are significantly greater than seen previously with RIIα and are likely to be important in mediating short range and long range interdomain and intersubunit interactions that uniquely regulate the activity of the type IIβ isoform of PKA.
Keywords: Cyclic AMP (cAMP); Intrinsically Disordered Protein; Protein Domain; Protein Dynamic; Protein Kinase A (PKA); Protein Structure; Small Angle Neutron Scattering (SANS); Small Angle X-ray Scattering (SAXS).
© 2014 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
Similar articles
-
Structure and allostery of the PKA RIIβ tetrameric holoenzyme.Science. 2012 Feb 10;335(6069):712-6. doi: 10.1126/science.1213979. Science. 2012. PMID: 22323819 Free PMC article.
-
Probing cAMP-dependent protein kinase holoenzyme complexes I alpha and II beta by FT-IR and chemical protein footprinting.Biochemistry. 2004 Feb 24;43(7):1908-20. doi: 10.1021/bi0354435. Biochemistry. 2004. PMID: 14967031
-
Structural analyses of the PKA RIIβ holoenzyme containing the oncogenic DnaJB1-PKAc fusion protein reveal protomer asymmetry and fusion-induced allosteric perturbations in fibrolamellar hepatocellular carcinoma.PLoS Biol. 2020 Dec 28;18(12):e3001018. doi: 10.1371/journal.pbio.3001018. eCollection 2020 Dec. PLoS Biol. 2020. PMID: 33370777 Free PMC article.
-
PKA: lessons learned after twenty years.Biochim Biophys Acta. 2013 Jul;1834(7):1271-8. doi: 10.1016/j.bbapap.2013.03.007. Epub 2013 Mar 25. Biochim Biophys Acta. 2013. PMID: 23535202 Free PMC article. Review.
-
Signaling through cAMP and cAMP-dependent protein kinase: diverse strategies for drug design.Biochim Biophys Acta. 2008 Jan;1784(1):16-26. doi: 10.1016/j.bbapap.2007.10.002. Epub 2007 Oct 12. Biochim Biophys Acta. 2008. PMID: 17996741 Free PMC article. Review.
Cited by
-
Mechanism of cAMP Partial Agonism in Protein Kinase G (PKG).J Biol Chem. 2015 Nov 27;290(48):28631-41. doi: 10.1074/jbc.M115.685305. Epub 2015 Sep 14. J Biol Chem. 2015. PMID: 26370085 Free PMC article.
References
-
- Soderling T. R., Hickenbottom J. P., Reimann E. M., Hunkeler F. L., Walsh D. A., Krebs E. G. (1970) Inactivation of glycogen synthetase and activation of phosphorylase kinase by muscle adenosine 3′,5′-monophosphate-dependent protein kinases. J. Biol. Chem. 245, 6317–6328 - PubMed
-
- McDonald T. F., Pelzer S., Trautwein W., Pelzer D. J. (1994) Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiol. Rev. 74, 365–507 - PubMed
-
- Wang J. Q., Liu X., Zhang G., Parelkar N. K., Arora A., Haines M., Fibuch E. E., Mao L. (2006) Phosphorylation of glutamate receptors: a potential mechanism for the regulation of receptor function and psychostimulant action. J. Neurosci. Res. 84, 1621–1629 - PubMed
-
- Shaywitz A. J., Greenberg M. E. (1999) CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu. Rev. Biochem. 68, 821–861 - PubMed
-
- Kirschner L. S., Carney J. A., Pack S. D., Taymans S. E., Giatzakis C., Cho Y. S., Cho-Chung Y. S., Stratakis C. A. (2000) Mutations of the gene encoding the protein kinase A type I-α regulatory subunit in patients with the Carney complex. Nat. Genet. 26, 89–92 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
