

Autosomal recessive polycystic kidney disease: a hepatorenal fibrocystic disorder with pleiotropic effects
- PMID: 25113295
- PMCID: PMC4143997
- DOI: 10.1542/peds.2013-3646
Autosomal recessive polycystic kidney disease: a hepatorenal fibrocystic disorder with pleiotropic effects
Abstract
Autosomal recessive polycystic kidney disease (ARPKD) is an important cause of chronic kidney disease in children. The care of ARPKD patients has traditionally been the realm of pediatric nephrologists; however, the disease has multisystem effects, and a comprehensive care strategy often requires a multidisciplinary team. Most notably, ARPKD patients have congenital hepatic fibrosis, which can lead to portal hypertension, requiring close follow-up by pediatric gastroenterologists. In severely affected infants, the diagnosis is often first suspected by obstetricians detecting enlarged, echogenic kidneys and oligohydramnios on prenatal ultrasounds. Neonatologists are central to the care of these infants, who may have respiratory compromise due to pulmonary hypoplasia and massively enlarged kidneys. Surgical considerations can include the possibility of nephrectomy to relieve mass effect, placement of dialysis access, and kidney and/or liver transplantation. Families of patients with ARPKD also face decisions regarding genetic testing of affected children, testing of asymptomatic siblings, or consideration of preimplantation genetic diagnosis for future pregnancies. They may therefore interface with genetic counselors, geneticists, and reproductive endocrinologists. Children with ARPKD may also be at risk for neurocognitive dysfunction and may require neuropsychological referral. The care of patients and families affected by ARPKD is therefore a multidisciplinary effort, and the general pediatrician can play a central role in this complex web of care. In this review, we outline the spectrum of clinical manifestations of ARPKD and review genetics of the disease, clinical and genetic diagnosis, perinatal management, management of organ-specific complications, and future directions for disease monitoring and potential therapies.
Keywords: congenital hepatic fibrosis; dialysis; genetic testing; kidney transplantation; liver transplantation; polycystic kidney disease; preimplantation genetic diagnosis.
Copyright © 2014 by the American Academy of Pediatrics.
Figures
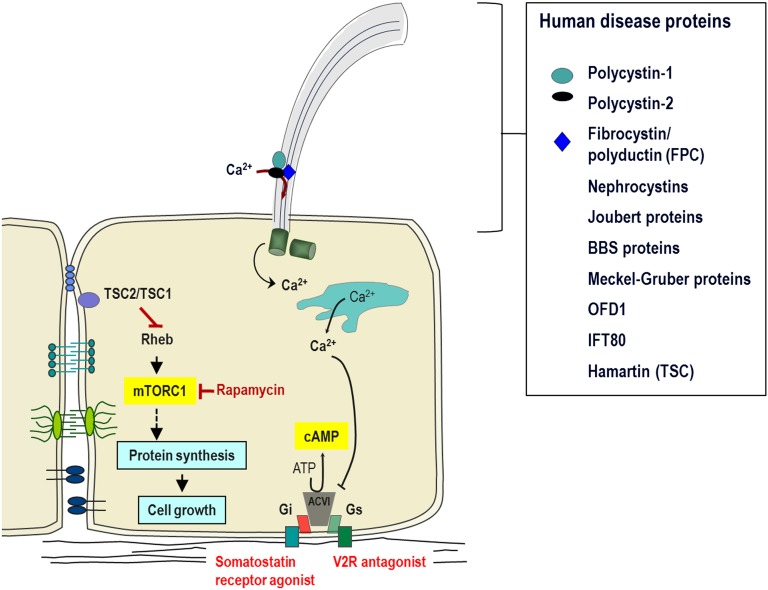
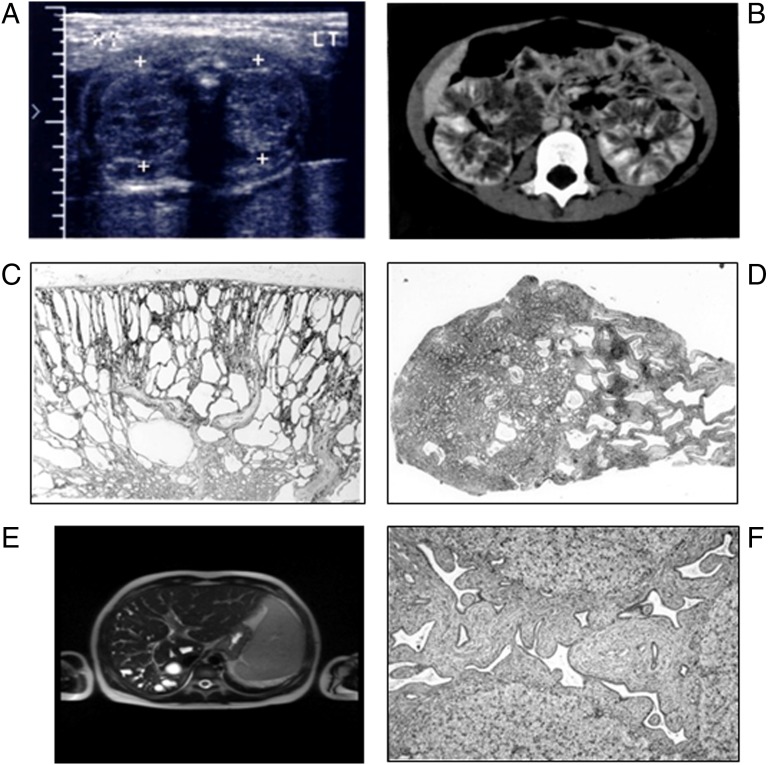
References
-
- Zerres K, Mücher G, Becker J, et al. . Prenatal diagnosis of autosomal recessive polycystic kidney disease (ARPKD): molecular genetics, clinical experience, and fetal morphology. Am J Med Genet. 1998;76(2):137–144 - PubMed
-
- Roy S, Dillon MJ, Trompeter RS, Barratt TM. Autosomal recessive polycystic kidney disease: long-term outcome of neonatal survivors. Pediatr Nephrol. 1997;11(3):302–306 - PubMed
-
- Kaplan BS, Fay J, Shah V, Dillon MJ, Barratt TM. Autosomal recessive polycystic kidney disease. Pediatr Nephrol. 1989;3(1):43–49 - PubMed
-
- Kääriäinen H, Jääskeläinen J, Kivisaari L, Koskimies O, Norio R. Dominant and recessive polycystic kidney disease in children: classification by intravenous pyelography, ultrasound, and computed tomography. Pediatr Radiol. 1988;18(1):45–50 - PubMed
-
- Adeva M, El-Youssef M, Rossetti S, et al. . Clinical and molecular characterization defines a broadened spectrum of autosomal recessive polycystic kidney disease (ARPKD). Medicine (Baltimore). 2006;85(1):1–21 doi:10.1097/01.md.0000200165.90373.9a - PubMed
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
