

Hellbender genome sequences shed light on genomic expansion at the base of crown salamanders
- PMID: 25115007
- PMCID: PMC4122941
- DOI: 10.1093/gbe/evu143
Hellbender genome sequences shed light on genomic expansion at the base of crown salamanders
Abstract
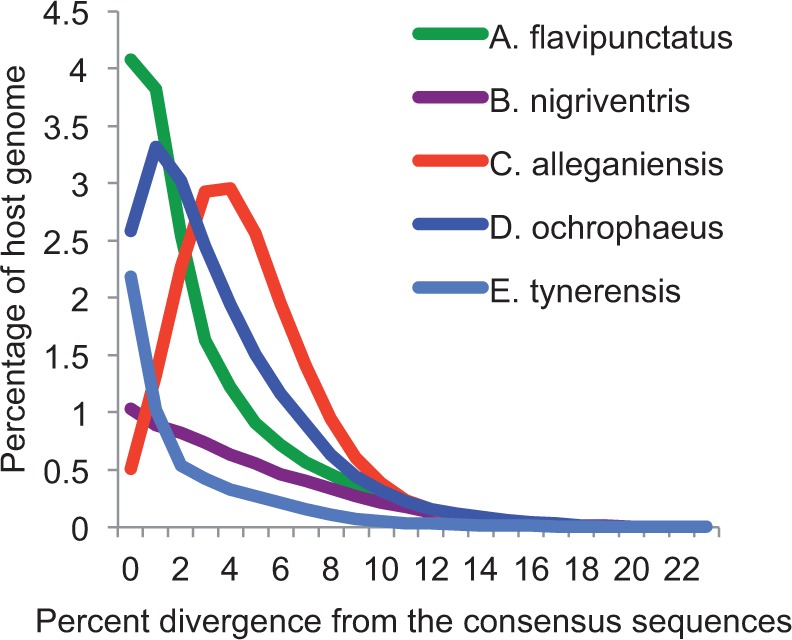
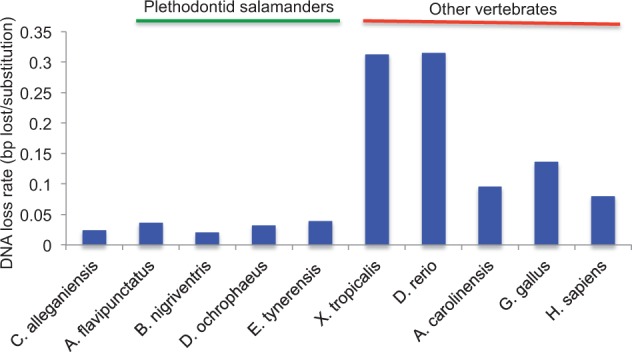
Among animals, genome sizes range from 20 Mb to 130 Gb, with 380-fold variation across vertebrates. Most of the largest vertebrate genomes are found in salamanders, an amphibian clade of 660 species. Thus, salamanders are an important system for studying causes and consequences of genomic gigantism. Previously, we showed that plethodontid salamander genomes accumulate higher levels of long terminal repeat (LTR) retrotransposons than do other vertebrates, although the evolutionary origins of such sequences remained unexplored. We also showed that some salamanders in the family Plethodontidae have relatively slow rates of DNA loss through small insertions and deletions. Here, we present new data from Cryptobranchus alleganiensis, the hellbender. Cryptobranchus and Plethodontidae span the basal phylogenetic split within salamanders; thus, analyses incorporating these taxa can shed light on the genome of the ancestral crown salamander lineage, which underwent expansion. We show that high levels of LTR retrotransposons likely characterize all crown salamanders, suggesting that disproportionate expansion of this transposable element (TE) class contributed to genomic expansion. Phylogenetic and age distribution analyses of salamander LTR retrotransposons indicate that salamanders' high TE levels reflect persistence and diversification of ancestral TEs rather than horizontal transfer events. Finally, we show that relatively slow DNA loss rates through small indels likely characterize all crown salamanders, suggesting that a decreased DNA loss rate contributed to genomic expansion at the clade's base. Our identification of shared genomic features across phylogenetically distant salamanders is a first step toward identifying the evolutionary processes underlying accumulation and persistence of high levels of repetitive sequence in salamander genomes.
Figures
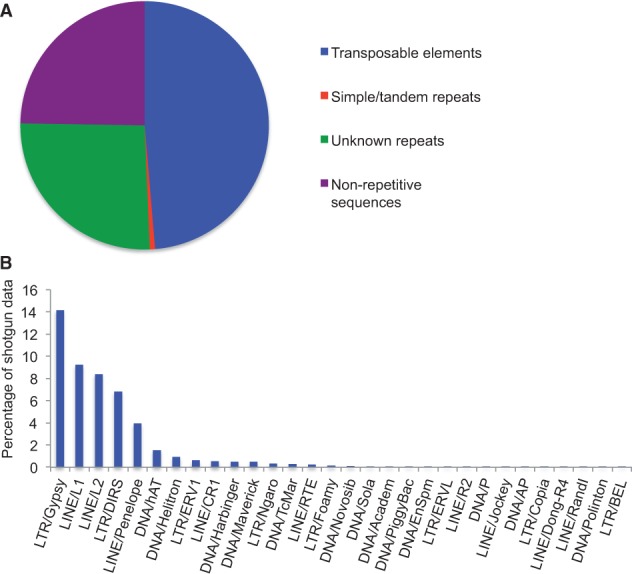
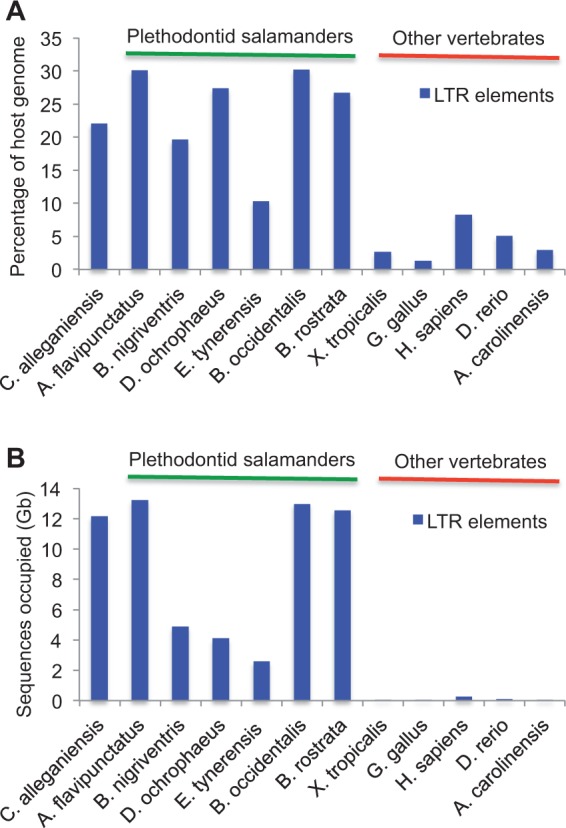
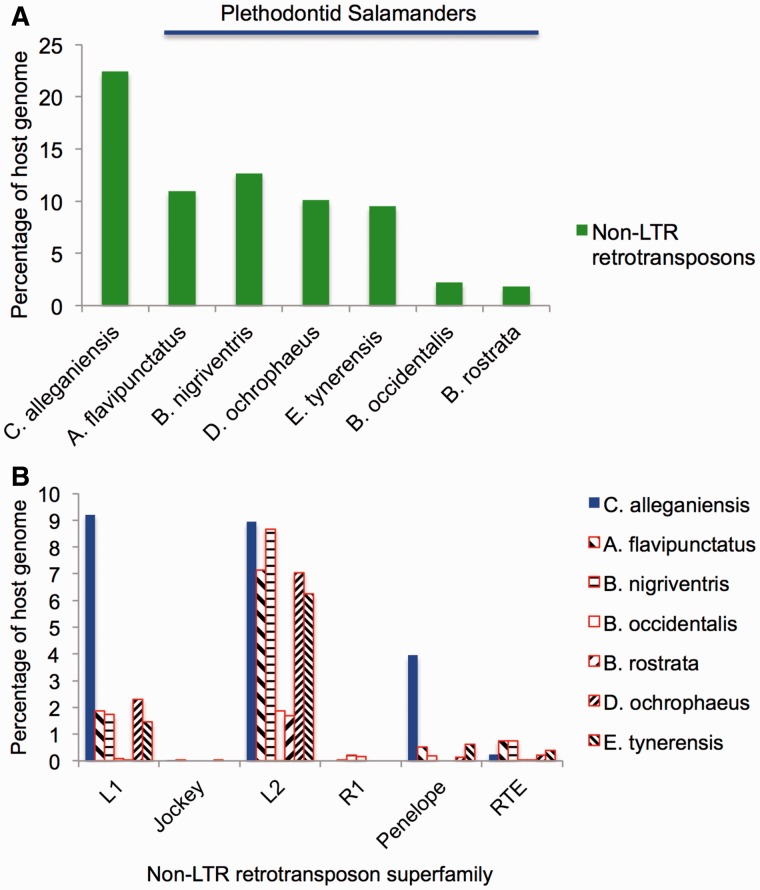
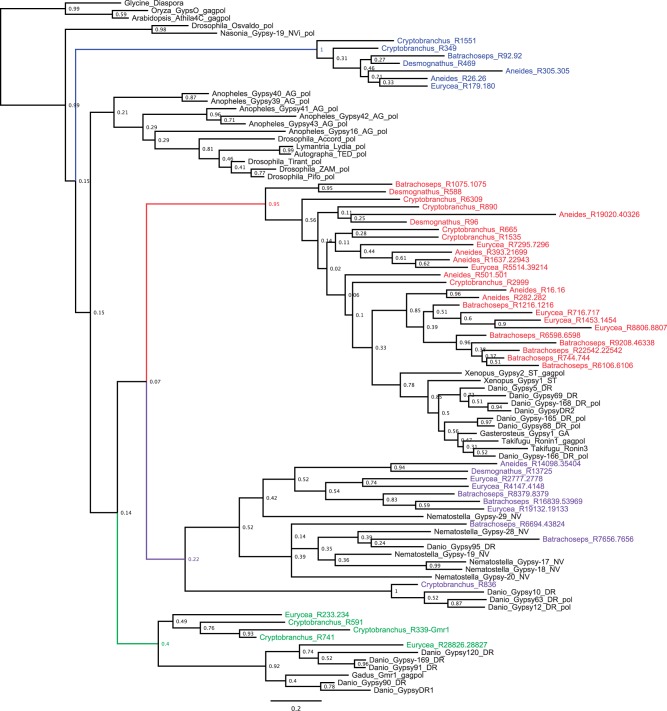
Similar articles
-
LTR retrotransposons contribute to genomic gigantism in plethodontid salamanders.Genome Biol Evol. 2012;4(2):168-83. doi: 10.1093/gbe/evr139. Epub 2011 Dec 26. Genome Biol Evol. 2012. PMID: 22200636 Free PMC article.
-
Low levels of LTR retrotransposon deletion by ectopic recombination in the gigantic genomes of salamanders.J Mol Evol. 2015 Feb;80(2):120-9. doi: 10.1007/s00239-014-9663-7. Epub 2015 Jan 22. J Mol Evol. 2015. PMID: 25608479
-
Slow DNA loss in the gigantic genomes of salamanders.Genome Biol Evol. 2012;4(12):1340-8. doi: 10.1093/gbe/evs103. Genome Biol Evol. 2012. PMID: 23175715 Free PMC article.
-
Co-evolution of plant LTR-retrotransposons and their host genomes.Protein Cell. 2013 Jul;4(7):493-501. doi: 10.1007/s13238-013-3037-6. Epub 2013 Jun 23. Protein Cell. 2013. PMID: 23794032 Free PMC article. Review.
-
Genome size, secondary simplification, and the evolution of the brain in salamanders.Brain Behav Evol. 1997 Jul;50(1):50-9. doi: 10.1159/000113321. Brain Behav Evol. 1997. PMID: 9209766 Review.
Cited by
-
Initial characterization of the large genome of the salamander Ambystoma mexicanum using shotgun and laser capture chromosome sequencing.Sci Rep. 2015 Nov 10;5:16413. doi: 10.1038/srep16413. Sci Rep. 2015. PMID: 26553646 Free PMC article.
-
Ecological constraints associated with genome size across salamander lineages.Proc Biol Sci. 2019 Sep 25;286(1911):20191780. doi: 10.1098/rspb.2019.1780. Epub 2019 Sep 18. Proc Biol Sci. 2019. PMID: 31530144 Free PMC article.
-
DNA gains and losses in gigantic genomes do not track differences in transposable element-host silencing interactions.Commun Biol. 2025 May 6;8(1):704. doi: 10.1038/s42003-025-08127-3. Commun Biol. 2025. PMID: 40328975 Free PMC article.
-
Virtual Genome Walking across the 32 Gb Ambystoma mexicanum genome; assembling gene models and intronic sequence.Sci Rep. 2018 Jan 12;8(1):618. doi: 10.1038/s41598-017-19128-6. Sci Rep. 2018. PMID: 29330416 Free PMC article.
-
Metamorphosis Imposes Variable Constraints on Genome Expansion through Effects on Development.Integr Org Biol. 2023 Apr 18;5(1):obad015. doi: 10.1093/iob/obad015. eCollection 2023. Integr Org Biol. 2023. PMID: 37143961 Free PMC article.
References
-
- AmphibiaWeb: information on amphibian biology and conservation [Internet] Berkeley (CA): AmphibiaWeb; 2014. [cited 2014 Apr 5]. Available from: http://amphibiaweb.org/
-
- Ball EV, et al. Microdeletions and microinsertions causing human genetic disease: common mechanisms of mutagenesis and the role of local DNA sequence complexity. Hum Mutat. 2005;26:205–213. - PubMed
-
- Batistoni R, Pesole G, Marracci S, Nardi I. A tandemly repeated DNA family originated from SINE-related elements in the European plethodontid salamanders (Amphibia, Urodela) J Mol Evol. 1995;40:608–615. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
