

Increased replicative fitness can lead to decreased drug sensitivity of hepatitis C virus
- PMID: 25122776
- PMCID: PMC4178724
- DOI: 10.1128/JVI.01860-14
Increased replicative fitness can lead to decreased drug sensitivity of hepatitis C virus
Abstract
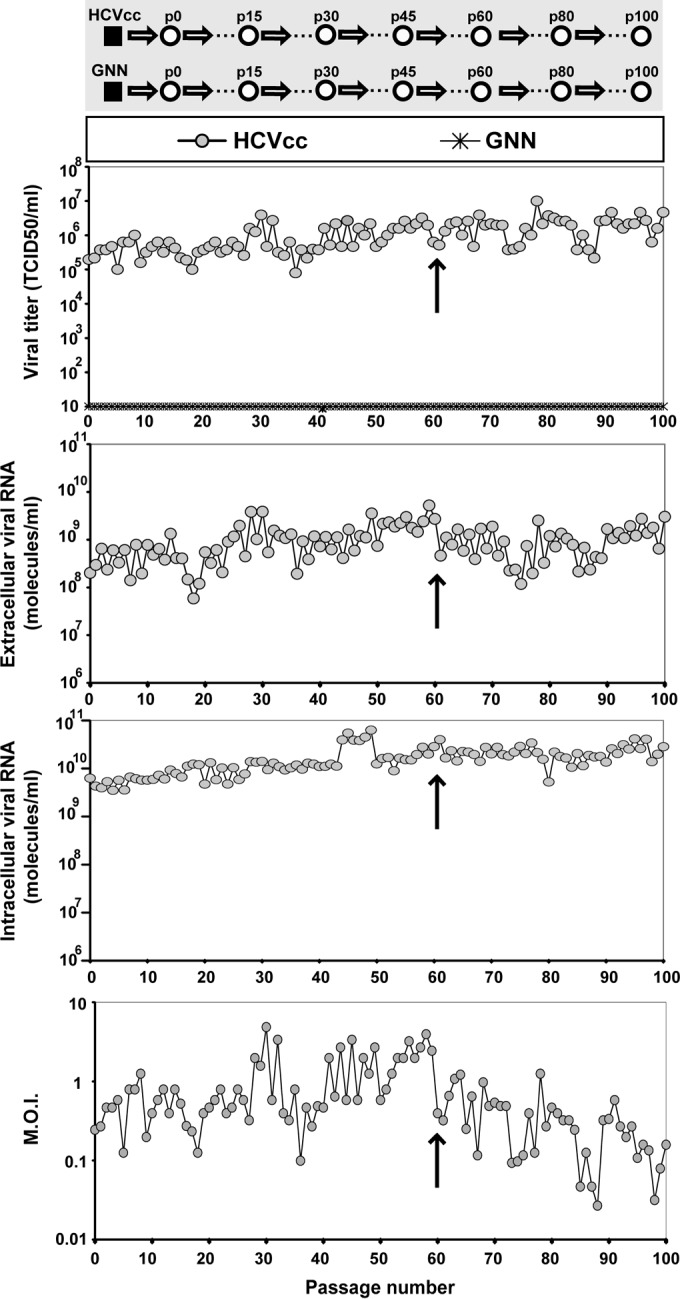
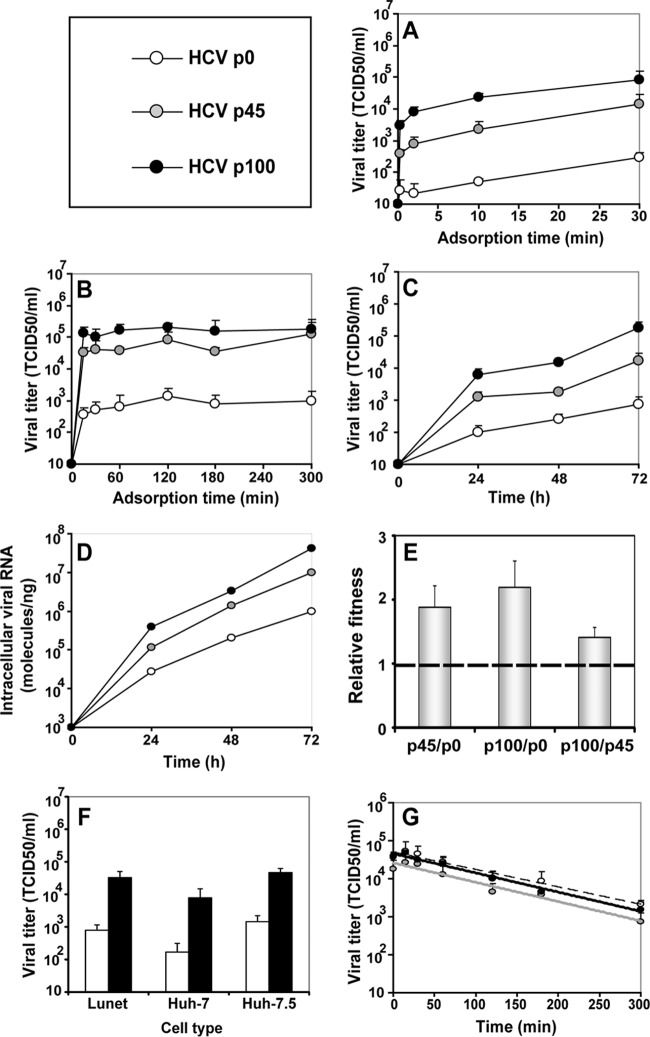
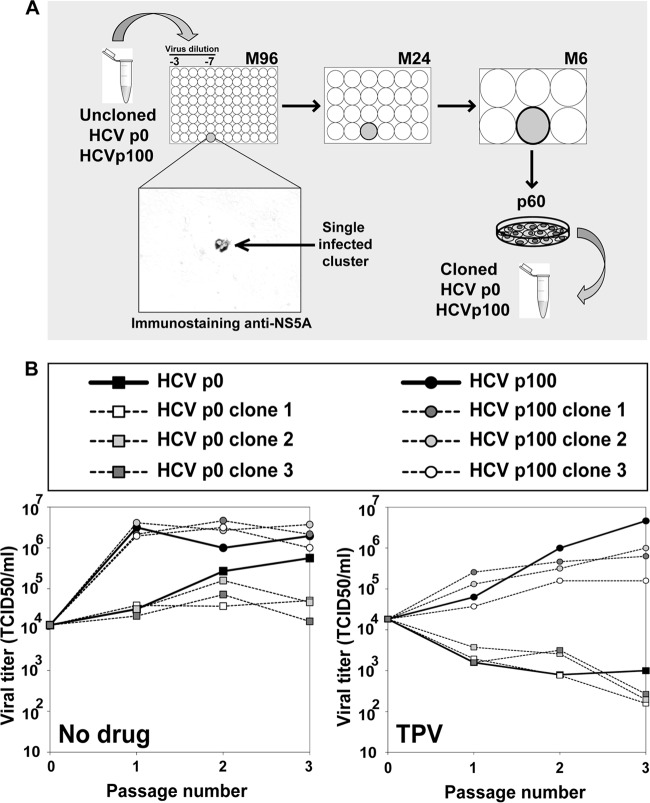
Passage of hepatitis C virus (HCV) in human hepatoma cells resulted in populations that displayed partial resistance to alpha interferon (IFN-α), telaprevir, daclatasvir, cyclosporine, and ribavirin, despite no prior exposure to these drugs. Mutant spectrum analyses and kinetics of virus production in the absence and presence of drugs indicate that resistance is not due to the presence of drug resistance mutations in the mutant spectrum of the initial or passaged populations but to increased replicative fitness acquired during passage. Fitness increases did not alter host factors that lead to shutoff of general host cell protein synthesis and preferential translation of HCV RNA. The results imply that viral replicative fitness is a mechanism of multidrug resistance in HCV. Importance: Viral drug resistance is usually attributed to the presence of amino acid substitutions in the protein targeted by the drug. In the present study with HCV, we show that high viral replicative fitness can confer a general drug resistance phenotype to the virus. The results exclude the possibility that genomes with drug resistance mutations are responsible for the observed phenotype. The fact that replicative fitness can be a determinant of multidrug resistance may explain why the virus is less sensitive to drug treatments in prolonged chronic HCV infections that favor increases in replicative fitness.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
Figures
References
-
- Suzuki F, Akuta N, Suzuki Y, Yatsuji H, Sezaki H, Arase Y, Kawamura Y, Hosaka T, Kobayashi M, Ikeda K, Kobayashi M, Watahiki S, Kumada H. 2007. Selection of a virus strain resistant to entecavir in a nucleoside-naive patient with hepatitis B of genotype H. J. Clin. Virol. 39:149–152. 10.1016/j.jcv.2007.03.004 - DOI - PubMed
-
- Domingo E. 1989. RNA virus evolution and the control of viral disease. Prog. Drug Res. 33:93–133 - PubMed
-
- Domingo E, Holland JJ. 1992. Complications of RNA heterogeneity for the engineering of virus vaccines and antiviral agents. Genet. Eng. (N Y) 14:13–31 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
