

Novel GALT variations and mutation spectrum in the Korean population with decreased galactose-1-phosphate uridyltransferase activity
- PMID: 25124065
- PMCID: PMC4236512
- DOI: 10.1186/s12881-014-0094-5
Novel GALT variations and mutation spectrum in the Korean population with decreased galactose-1-phosphate uridyltransferase activity
Abstract
Background: Classic galactosemia (OMIM #230400) is an autosomal recessive metabolic disorder caused by a deficiency of the galactose-1-phosphate uridyltransferase (GALT, EC2.7.7.12) protein due to mutations in the GALT gene. The aim of this study was to provide a comprehensive and updated mutation spectrum of GALT in a Korean population.
Methods: Thirteen unrelated patients screened positive for galactosemia in a newborn screening program were included in this study. They showed a reduced GALT enzyme activity in red blood cells. Direct sequencing of the GALT gene and in silico analyses were done to evaluate the impact of novel variations upon GALT enzyme activity. We also reviewed previous reports for GALT mutations in Koreans.
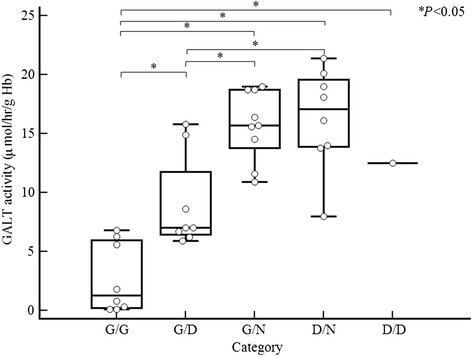
Results: We identified six novel likely pathogenic variations including three missense (p.Ala101Asp, p.Tyr165His, and p.Pro257Thr), one small deletion/insertion [c.826_827delinsAA (p.Ala276Asn)], one frameshift (p.Asn96Serfs*5), and one splicing (c.378-1G > C) likely pathogenic variations. The most frequent variation was the Duarte variant (c.940A > G, 35.3%), followed by c.507G > C (p.Gln169His, 9.6%), among 34 Korean patients. Other mutations were widely scattered. None of the eight common mutations used for targeted mutation analysis in Western countries including p.Gln188Arg, p.Ser135Leu, p.Lys285Asn, p.Leu195Pro, p.Tyr209Cys, p.Phe171Ser, c.253-2A > G, and a 5 kb deletion, had been found in Koreans until this study.
Conclusions: Considering the mutation spectrum in Koreans, direct sequence analysis of entire GALT exons is recommended for accurate diagnosis. The mutations responsible for GALT deficiency in the Korean population were clearly different from those of other populations.
Figures
References
-
- Waisbren SE, Potter NL, Gordon CM, Green RC, Greenstein P, Gubbels CS, Rubio-Gozalbo E, Schomer D, Welt C, Anastasoaie V, D’Anna K, Gentile J, Guo CY, Hecht L, Jackson R, Jansma BM, Li Y, Lip V, Miller DT, Murray M, Power L, Quinn N, Rohr F, Shen Y, Skinder-Meredith A, Timmers I, Tunick R, Wessel A, Wu BL, Levy H. The adult galactosemic phenotype. J Inherit Metab Dis. 2012;35(2):279–286. doi: 10.1007/s10545-011-9372-y. - DOI - PMC - PubMed
-
- Jumbo-Lucioni PP, Garber K, Kiel J, Baric I, Berry GT, Bosch A, Burlina A, Chiesa A, Pico ML, Estrada SC, Henderson H, Leslie N, Longo N, Morris AA, Ramirez-Farias C, Schweitzer-Krantz S, Silao CL, Vela-Amieva M, Waisbren S, Fridovich-Keil JL. Diversity of approaches to classic galactosemia around the world: a comparison of diagnosis, intervention, and outcomes. J Inherit Metab Dis. 2012;35(6):1037–1049. doi: 10.1007/s10545-012-9477-y. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
