

Finding the sweet spot: assembly and glycosylation of the dystrophin-associated glycoprotein complex
- PMID: 25125182
- PMCID: PMC4135523
- DOI: 10.1002/ar.22974
Finding the sweet spot: assembly and glycosylation of the dystrophin-associated glycoprotein complex
Abstract
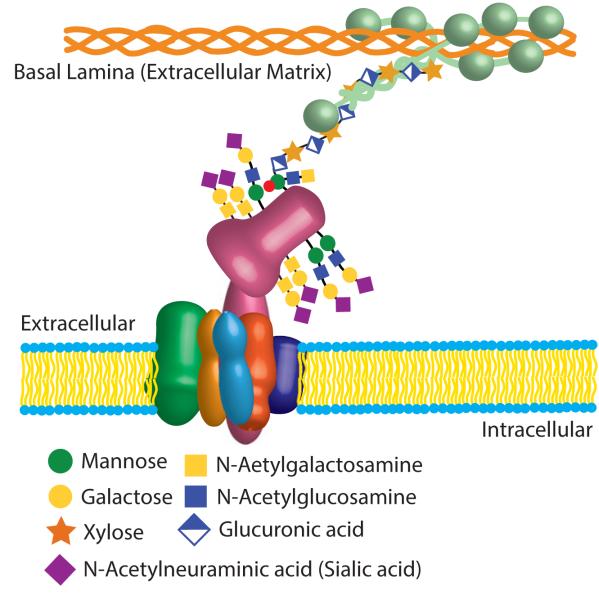
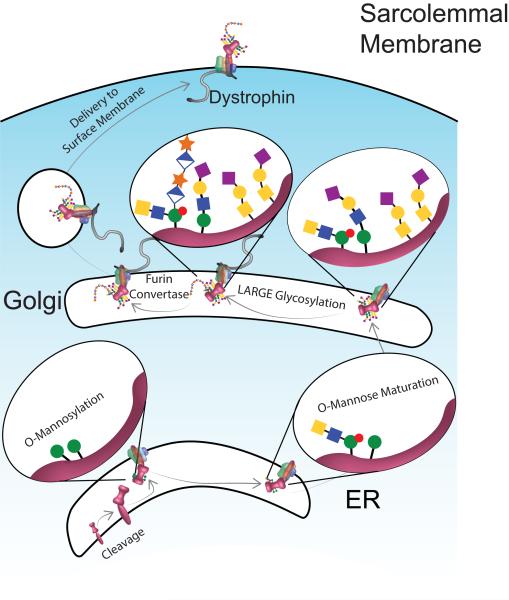
The dystrophin-associated glycoprotein complex (DGC) is a collection of glycoproteins that are essential for the normal function of striated muscle and many other tissues. Recent genetic studies have implicated the components of this complex in over a dozen forms of muscular dystrophy. Furthermore, disruption of the DGC has been implicated in many forms of acquired disease. This review aims to summarize the current state of knowledge regarding the processing and assembly of dystrophin-associated proteins with a focus primarily on the dystroglycan heterodimer and the sarcoglycan complex. These proteins form the transmembrane portion of the DGC and undergo a complex multi-step processing with proteolytic cleavage, differential assembly, and both N- and O-glycosylation. The enzymes responsible for this processing and a model describing the sequence and subcellular localization of these events are discussed.
Keywords: dystroglycan; dystrophin associated glycoprotein complexes; muscle; sarcoglycan; sarcospan.
© 2014 Wiley Periodicals, Inc.
Figures
References
-
- Acharyya S, Butchbach MER, Sahenk Z, Wang H, Saji M, Carathers M, Ringel MD, Skipworth RJE, Fearon KCH, Hollingsworth MA, Muscarella P, Burghes AHM, Rafael-Fortney JA, Guttridge DC. Dystrophin glycoprotein complex dysfunction: a regulatory link between muscular dystrophy and cancer cachexia. Cancer Cell. 2005;8:421–432. - PubMed
-
- Albrecht DE, Froehner SC. DAMAGE, a novel alpha-dystrobrevin-associated MAGE protein in dystrophin complexes. J Biol Chem. 2004;279:7014–7023. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
