

Genetic heterogeneity in Cornelia de Lange syndrome (CdLS) and CdLS-like phenotypes with observed and predicted levels of mosaicism
- PMID: 25125236
- PMCID: PMC4173748
- DOI: 10.1136/jmedgenet-2014-102573
Genetic heterogeneity in Cornelia de Lange syndrome (CdLS) and CdLS-like phenotypes with observed and predicted levels of mosaicism
Abstract
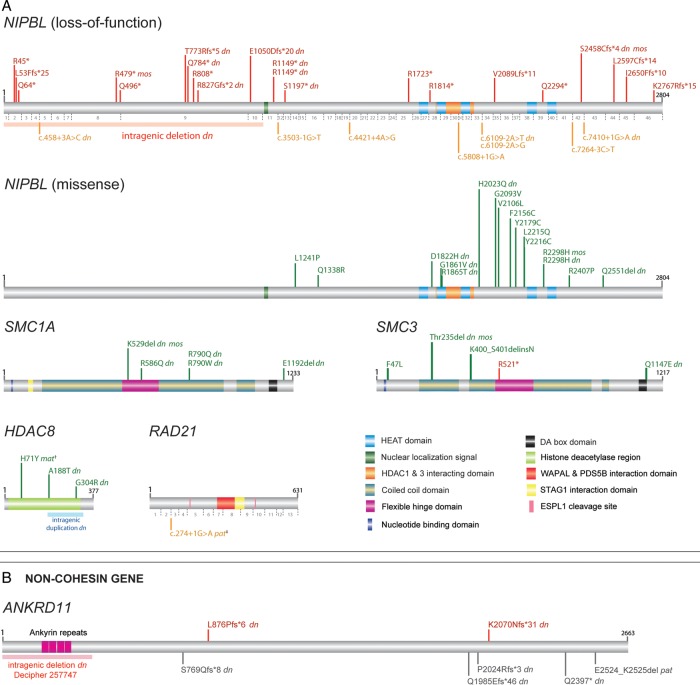
Background: Cornelia de Lange syndrome (CdLS) is a multisystem disorder with distinctive facial appearance, intellectual disability and growth failure as prominent features. Most individuals with typical CdLS have de novo heterozygous loss-of-function mutations in NIPBL with mosaic individuals representing a significant proportion. Mutations in other cohesin components, SMC1A, SMC3, HDAC8 and RAD21 cause less typical CdLS.
Methods: We screened 163 affected individuals for coding region mutations in the known genes, 90 for genomic rearrangements, 19 for deep intronic variants in NIPBL and 5 had whole-exome sequencing.
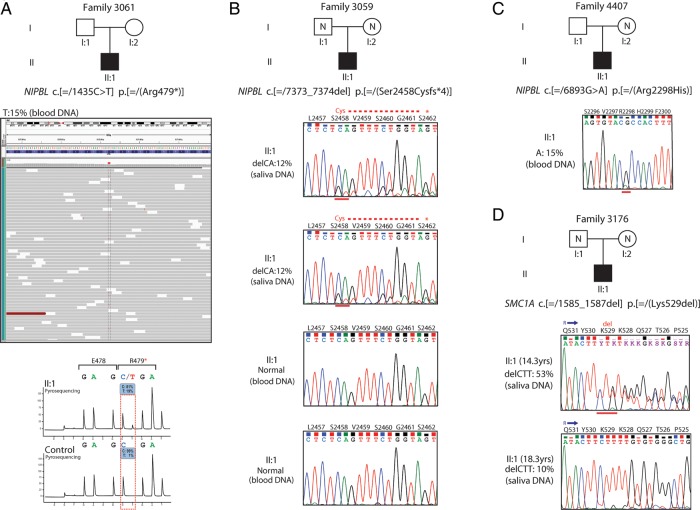
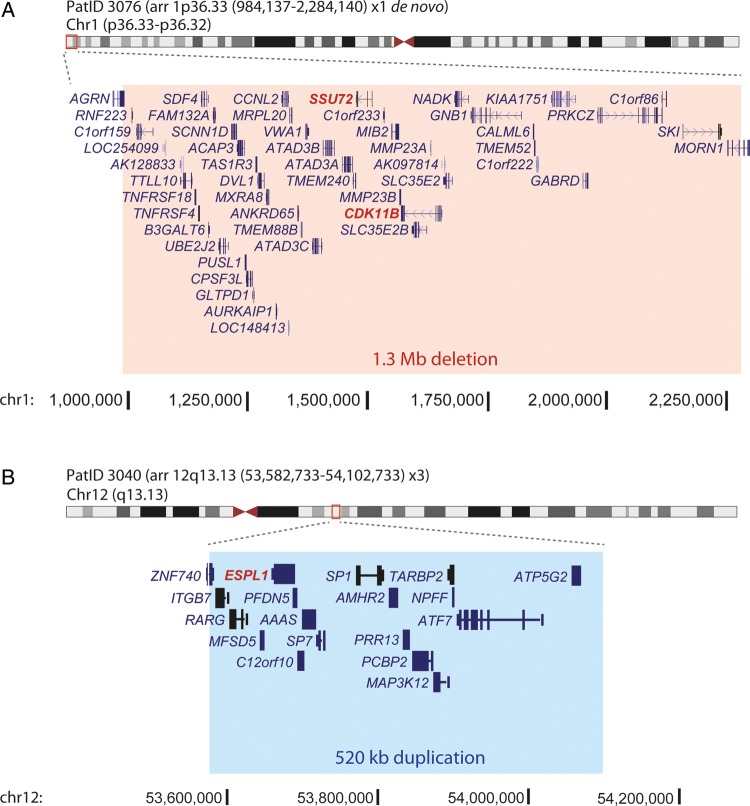
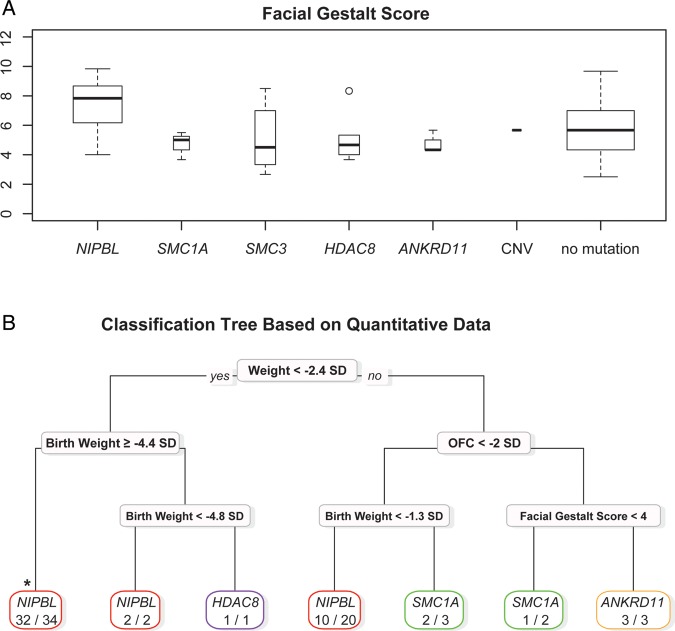
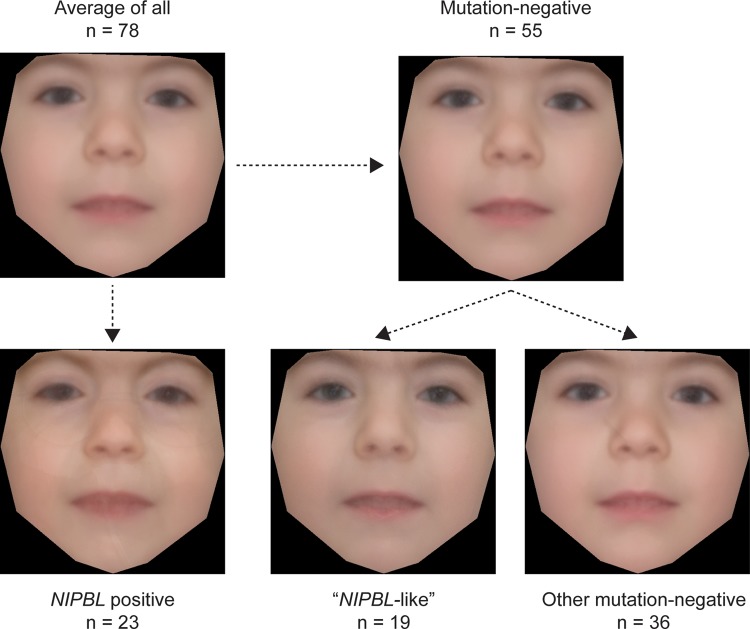
Results: Pathogenic mutations [including mosaic changes] were identified in: NIPBL 46 [3] (28.2%); SMC1A 5 [1] (3.1%); SMC3 5 [1] (3.1%); HDAC8 6 [0] (3.6%) and RAD21 1 [0] (0.6%). One individual had a de novo 1.3 Mb deletion of 1p36.3. Another had a 520 kb duplication of 12q13.13 encompassing ESPL1, encoding separase, an enzyme that cleaves the cohesin ring. Three de novo mutations were identified in ANKRD11 demonstrating a phenotypic overlap with KBG syndrome. To estimate the number of undetected mosaic cases we used recursive partitioning to identify discriminating features in the NIPBL-positive subgroup. Filtering of the mutation-negative group on these features classified at least 18% as 'NIPBL-like'. A computer composition of the average face of this NIPBL-like subgroup was also more typical in appearance than that of all others in the mutation-negative group supporting the existence of undetected mosaic cases.
Conclusions: Future diagnostic testing in 'mutation-negative' CdLS thus merits deeper sequencing of multiple DNA samples derived from different tissues.
Keywords: Clinical genetics; Copy-number; Molecular genetics.
Published by the BMJ Publishing Group Limited. For permission to use (where not already granted under a licence) please go to http://group.bmj.com/group/rights-licensing/permissions.
Figures
References
-
- Kline AD, Krantz ID, Sommer A, Kliewer M, Jackson LG, FitzPatrick DR, Levin AV, Selicorni A. Cornelia de Lange syndrome: clinical review, diagnostic and scoring systems, and anticipatory guidance. Am J Med Genet A 2007;143A:1287–96 - PubMed
-
- Krantz ID, McCallum J, DeScipio C, Kaur M, Gillis LA, Yaeger D, Jukofsky L, Wasserman N, Bottani A, Morris CA, Nowaczyk MJM, Toriello H, Bamshad MJ, Carey JC, Rappaport E, Kawauchi S, Lander AD, Calof AL, Li HH, Devoto M, Jackson LG. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat Genet 2004;36:631–5 - PMC - PubMed
-
- Tonkin ET, Wang TJ, Lisgo S, Bamshad MJ, Strachan T. NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat Genet 2004;36:636–41 - PubMed
-
- Baquero-Montoya C, Gil-Rodriguez MC, Braunholz D, Teresa-Rodrigo ME, Obieglo C, Gener B, Schwarzmayr T, Strom TM, Gomez-Puertas P, Puisac B, Gillessen-Kaesbach G, Musio A, Ramos FJ, Kaiser FJ, Pie J. Somatic mosaicism in a Cornelia de Lange syndrome patient with NIPBL mutation identified by different next generation sequencing approaches. Clin Genet 2014; 10.1111 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous