

Endoplasmic reticulum stress induced by tunicamycin and thapsigargin protects against transient ischemic brain injury: Involvement of PARK2-dependent mitophagy
- PMID: 25126734
- PMCID: PMC4198364
- DOI: 10.4161/auto.32136
Endoplasmic reticulum stress induced by tunicamycin and thapsigargin protects against transient ischemic brain injury: Involvement of PARK2-dependent mitophagy
Abstract
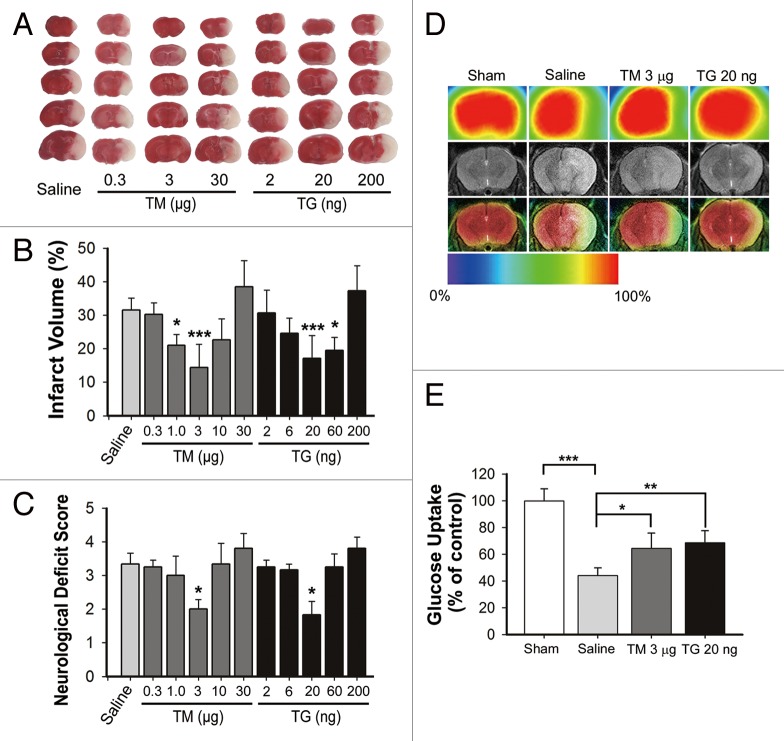
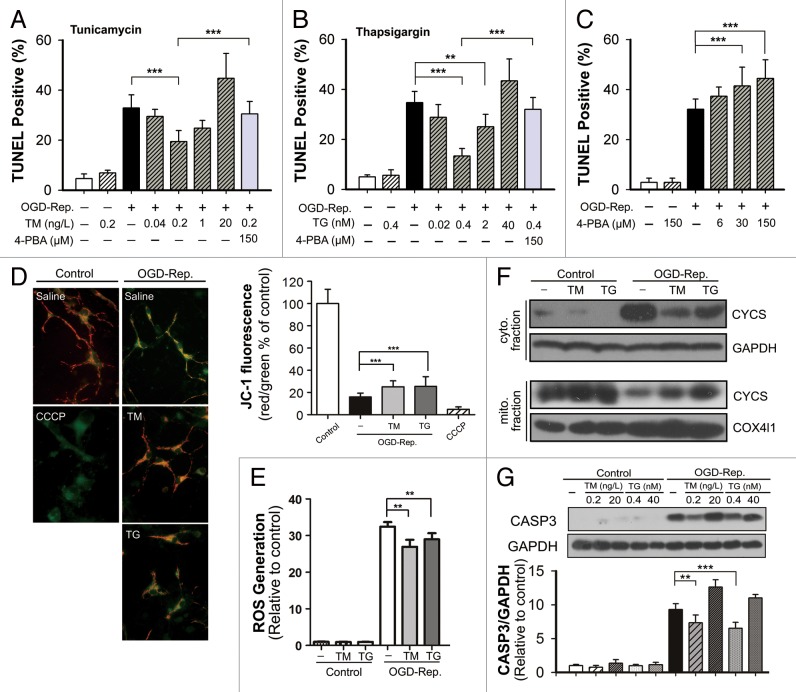
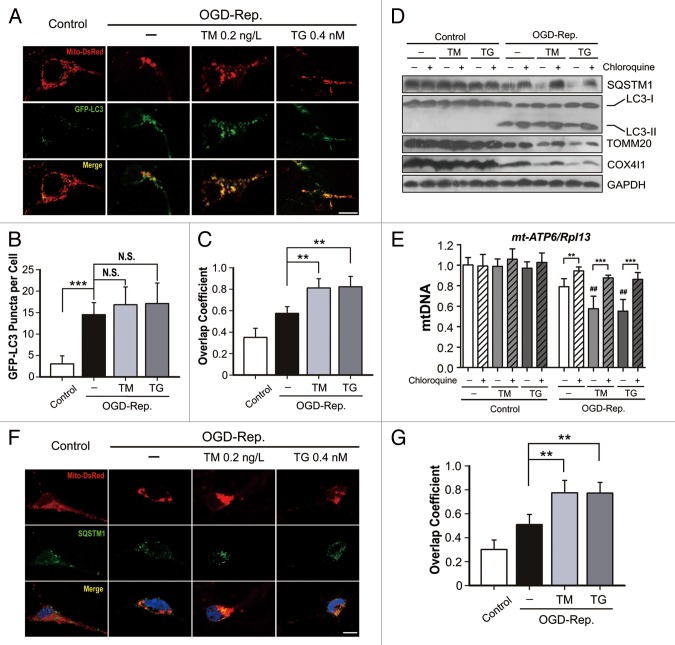
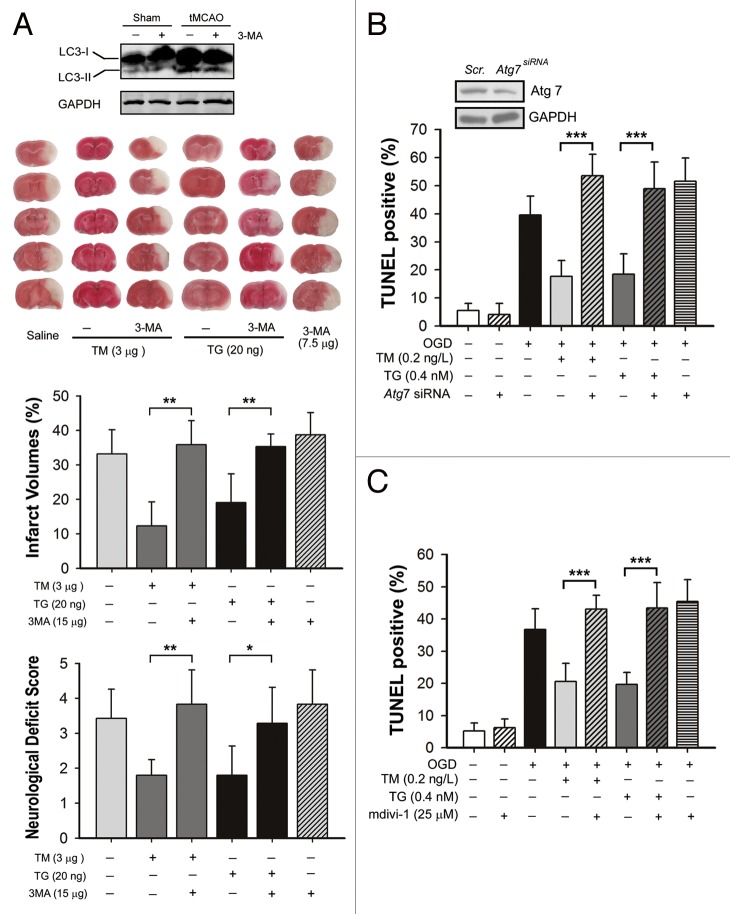
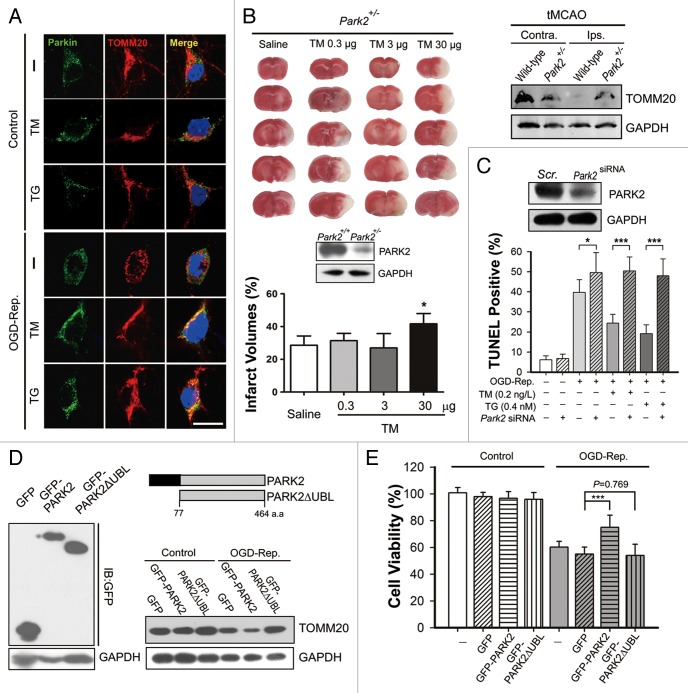
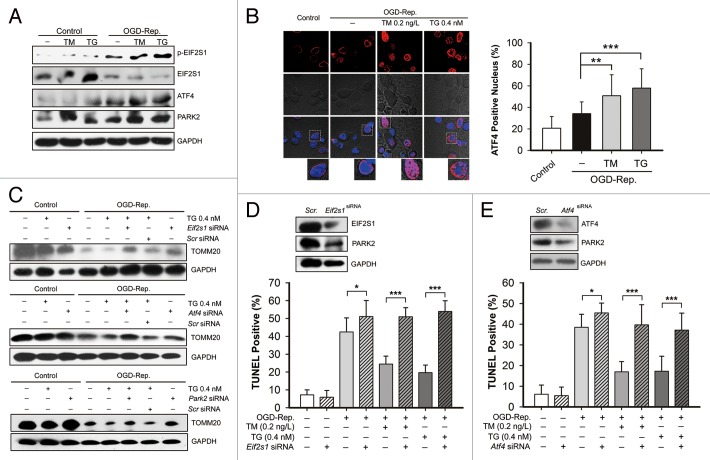
Transient cerebral ischemia leads to endoplasmic reticulum (ER) stress. However, the contributions of ER stress to cerebral ischemia are not clear. To address this issue, the ER stress activators tunicamycin (TM) and thapsigargin (TG) were administered to transient middle cerebral artery occluded (tMCAO) mice and oxygen-glucose deprivation-reperfusion (OGD-Rep.)-treated neurons. Both TM and TG showed significant protection against ischemia-induced brain injury, as revealed by reduced brain infarct volume and increased glucose uptake rate in ischemic tissue. In OGD-Rep.-treated neurons, 4-PBA, the ER stress releasing mechanism, counteracted the neuronal protection of TM and TG, which also supports a protective role of ER stress in transient brain ischemia. Knocking down the ER stress sensor Eif2s1, which is further activated by TM and TG, reduced the OGD-Rep.-induced neuronal cell death. In addition, both TM and TG prevented PARK2 loss, promoted its recruitment to mitochondria, and activated mitophagy during reperfusion after ischemia. The neuroprotection of TM and TG was reversed by autophagy inhibition (3-methyladenine and Atg7 knockdown) as well as Park2 silencing. The neuroprotection was also diminished in Park2(+/-) mice. Moreover, Eif2s1 and downstream Atf4 silencing reduced PARK2 expression, impaired mitophagy induction, and counteracted the neuroprotection. Taken together, the present investigation demonstrates that the ER stress induced by TM and TG protects against the transient ischemic brain injury. The PARK2-mediated mitophagy may be underlying the protection of ER stress. These findings may provide a new strategy to rescue ischemic brains by inducing mitophagy through ER stress activation.
Keywords: PARK2; cerebral ischemia; endoplasmic reticulum stress; mitophagy; neuroprotection.
Figures
References
-
- Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A. 2005;102:12005–10. doi: 10.1073/pnas.0505294102. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous