

Longevity of patients with cystic fibrosis in 2000 to 2010 and beyond: survival analysis of the Cystic Fibrosis Foundation patient registry
- PMID: 25133359
- PMCID: PMC4687404
- DOI: 10.7326/M13-0636
Longevity of patients with cystic fibrosis in 2000 to 2010 and beyond: survival analysis of the Cystic Fibrosis Foundation patient registry
Abstract
Background: Advances in treatments for cystic fibrosis (CF) continue to extend survival. An updated estimate of survival is needed for better prognostication and to anticipate evolving adult care needs.
Objective: To characterize trends in CF survival between 2000 and 2010 and to project survival for children born and diagnosed with the disease in 2010.
Design: Registry-based study.
Setting: 110 Cystic Fibrosis Foundation-accredited care centers in the United States.
Patients: All patients represented in the Cystic Fibrosis Foundation Patient Registry (CFFPR) between 2000 and 2010.
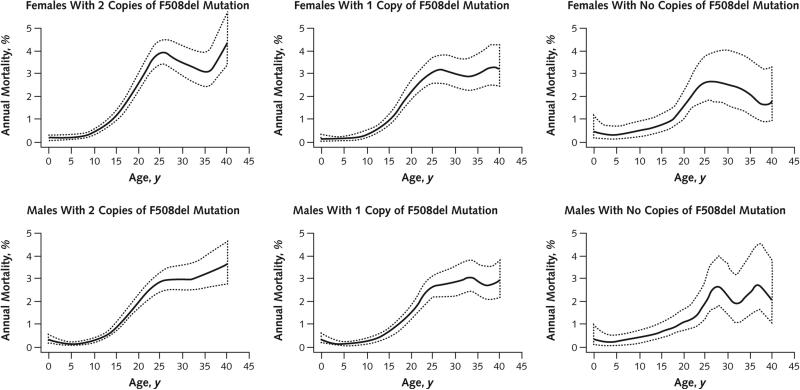
Measurements: Survival was modeled with respect to age, age at diagnosis, gender, race or ethnicity, F508del mutation status, and symptoms at diagnosis.
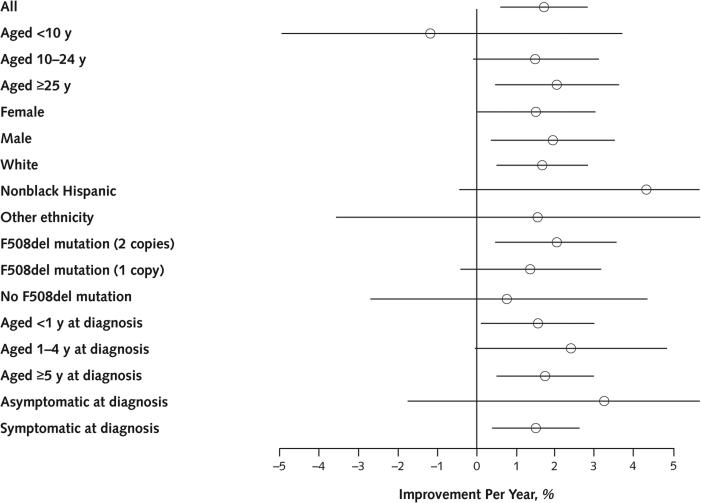
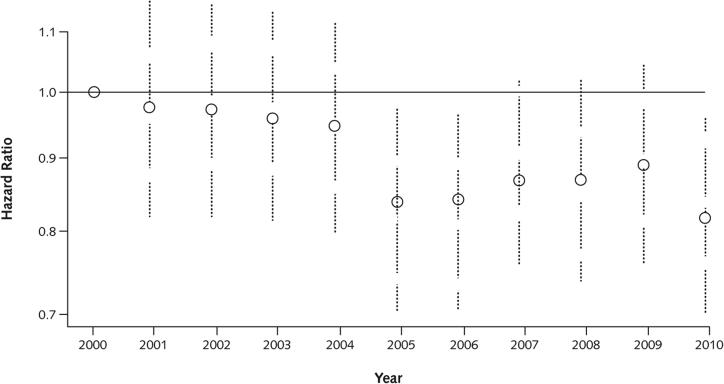
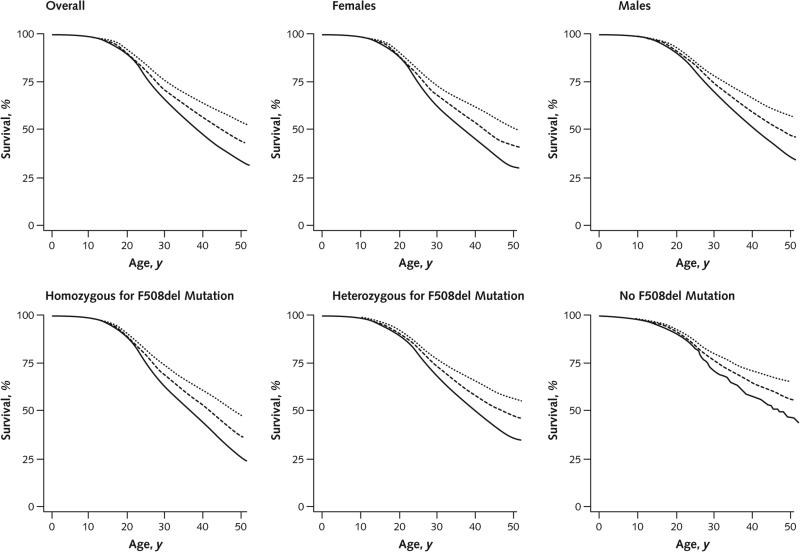
Results: Between 2000 and 2010, the number of patients in the CFFPR increased from 21,000 to 26,000, median age increased from 14.3 to 16.7 years, and adjusted mortality decreased by 1.8% per year (95% CI, 0.5% to 2.7%). Males had a 19% (CI, 13% to 24%) lower adjusted risk for death than females. Median survival of children born and diagnosed with CF in 2010 is projected to be 37 years (CI, 35 to 39 years) for females and 40 years (CI, 39 to 42 years) for males if mortality remains at 2010 levels and more than 50 years if mortality continues to decrease at the rate observed between 2000 and 2010.
Limitations: The CFFPR does not include all patients with CF in the United States, and loss to follow-up and missing data were observed. Additional analyses to address these limitations suggest that the survival projections are conservative.
Conclusion: Children born and diagnosed with CF in the United States in 2010 are expected to live longer than those born earlier. This has important implications for prognostic discussions and suggests that the health care system should anticipate greater numbers of adults with CF.
Primary funding source: Cystic Fibrosis Foundation.
Figures
Comment in
-
Cystic fibrosis: recent successes present new challenges.Ann Intern Med. 2014 Aug 19;161(4):298-9. doi: 10.7326/M14-1534. Ann Intern Med. 2014. PMID: 25133363 No abstract available.
Summary for patients in
-
Summaries for patients. Improved survival of patients with cystic fibrosis.Ann Intern Med. 2014 Aug 19;161(4):I-15. doi: 10.7326/P14-9026. Ann Intern Med. 2014. PMID: 25133375 No abstract available.
References
-
- Cystic Fibrosis Foundation . About Cystic Fibrosis. Cystic Fibrosis Foundation; Bethesda, MD: 2012. [12 December 2012]. Accessed at www.cff.org/AboutCF.
-
- FitzSimmons SC. The changing epidemiology of cystic fibrosis. J Pediatr. 1993;122:1–9. [PMID: 8419592] - PubMed
-
- Mayer-Hamblett N, Rosenfeld M, Emerson J, Goss CH, Aitken ML. Developing cystic fibrosis lung transplant referral criteria using predictors of 2-year mortality. Am J Respir Crit Care Med. 2002;166:1550–5. [PMID: 12406843] - PubMed
-
- Dasenbrook EC, Checkley W, Merlo CA, Konstan MW, Lechtzin N, Boyle MP. Association between respiratory tract methicillin-resistant Staphylococcus aureus and survival in cystic fibrosis. JAMA. 2010;303:2386–92. [PMID: 20551409] - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical