

FANCD2-controlled chromatin access of the Fanconi-associated nuclease FAN1 is crucial for the recovery of stalled replication forks
- PMID: 25135477
- PMCID: PMC4386448
- DOI: 10.1128/MCB.00457-14
FANCD2-controlled chromatin access of the Fanconi-associated nuclease FAN1 is crucial for the recovery of stalled replication forks
Abstract
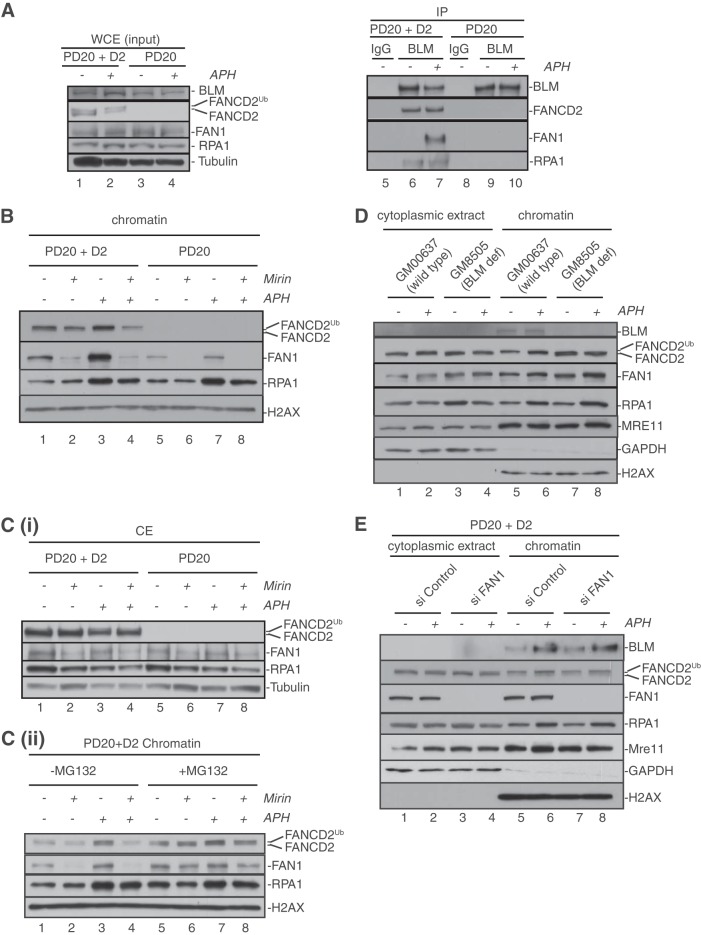
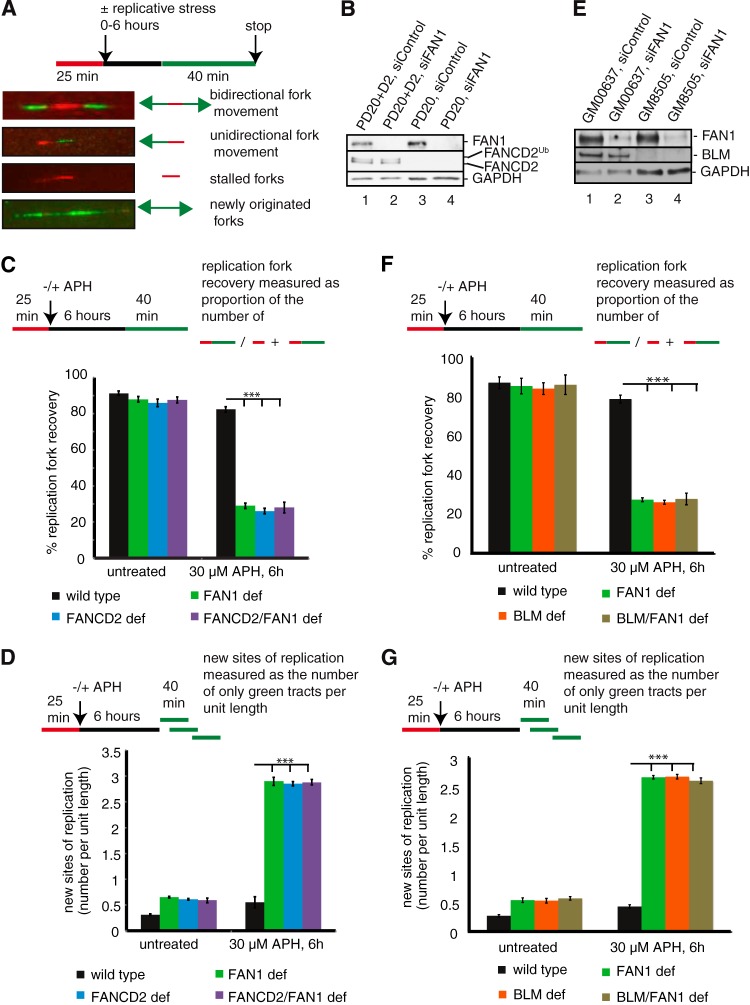
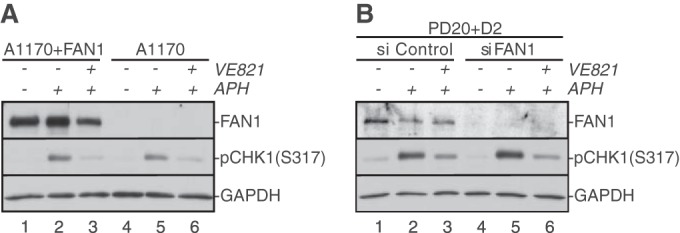
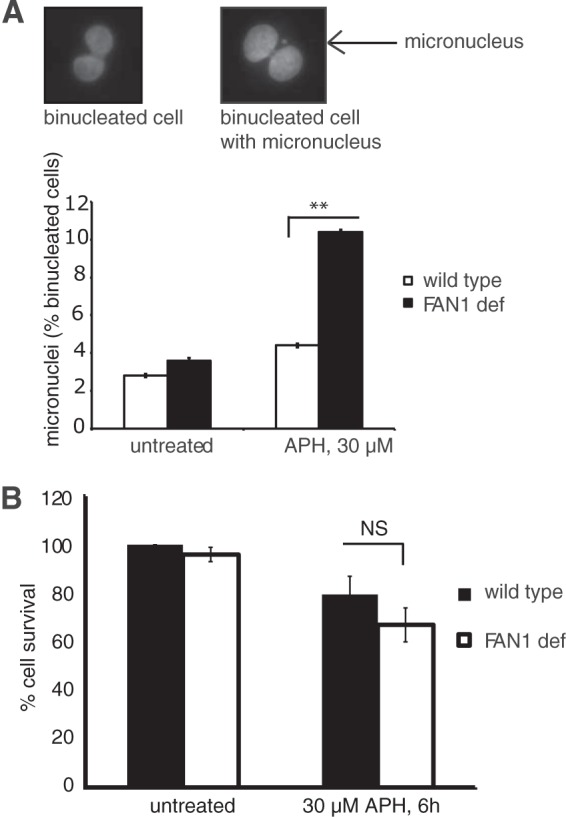
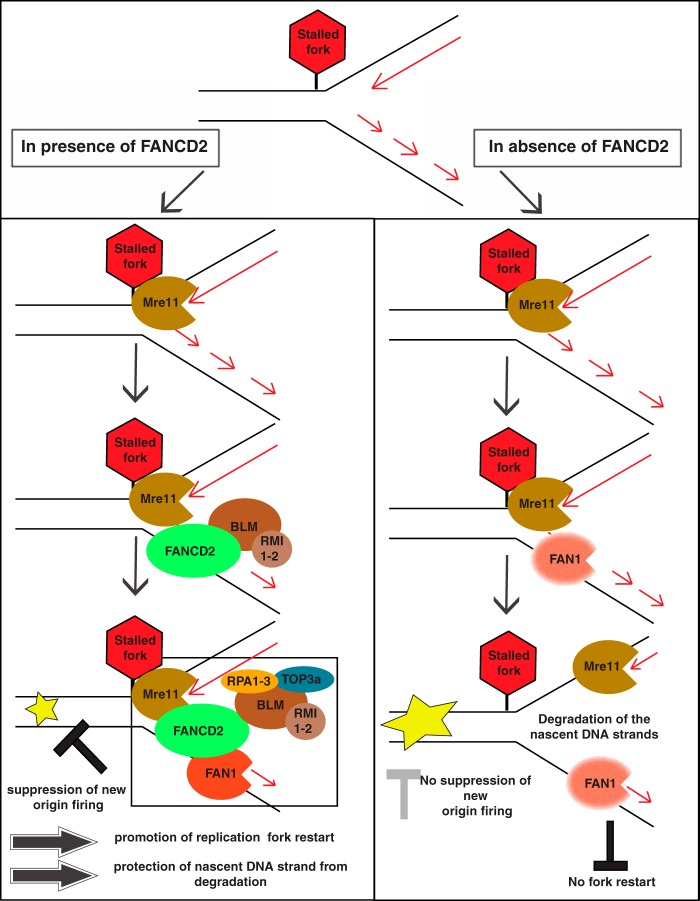
Fanconi anemia (FA) is a cancer predisposition syndrome characterized by cellular hypersensitivity to DNA interstrand cross-links (ICLs). Within the FA pathway, an upstream core complex monoubiquitinates and recruits the FANCD2 protein to ICLs on chromatin. Ensuing DNA repair involves the Fanconi-associated nuclease 1 (FAN1), which interacts selectively with monoubiquitinated FANCD2 (FANCD2(Ub)) at ICLs. Importantly, FANCD2 has additional independent functions: it binds chromatin and coordinates the restart of aphidicolin (APH)-stalled replication forks in concert with the BLM helicase, while protecting forks from nucleolytic degradation by MRE11. We identified FAN1 as a new crucial replication fork recovery factor. FAN1 joins the BLM-FANCD2 complex following APH-mediated fork stalling in a manner dependent on MRE11 and FANCD2, followed by FAN1 nuclease-mediated fork restart. Surprisingly, APH-induced activation and chromatin recruitment of FAN1 occur independently of the FA core complex or the FAN1 UBZ domain, indicating that the FANCD2(Ub) isoform is dispensable for functional FANCD2-FAN1 cross talk during stalled fork recovery. In the absence of FANCD2, MRE11 exonuclease-promoted access of FAN1 to stalled forks results in severe FAN1-mediated nucleolytic degradation of nascent DNA strands. Thus, FAN1 nuclease activity at stalled replication forks requires tight regulation: too little inhibits fork restart, whereas too much causes fork degradation.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
Figures
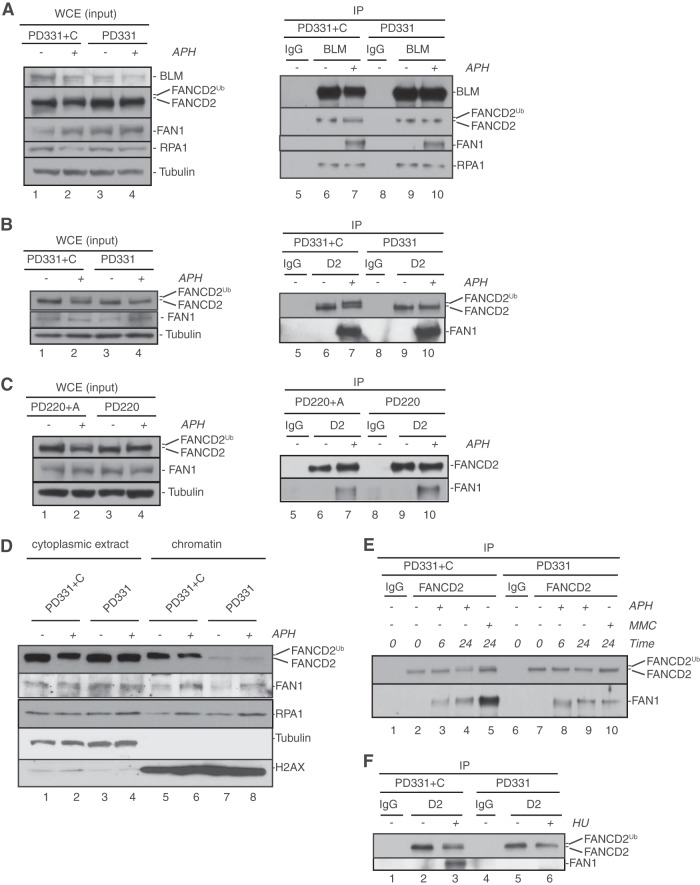
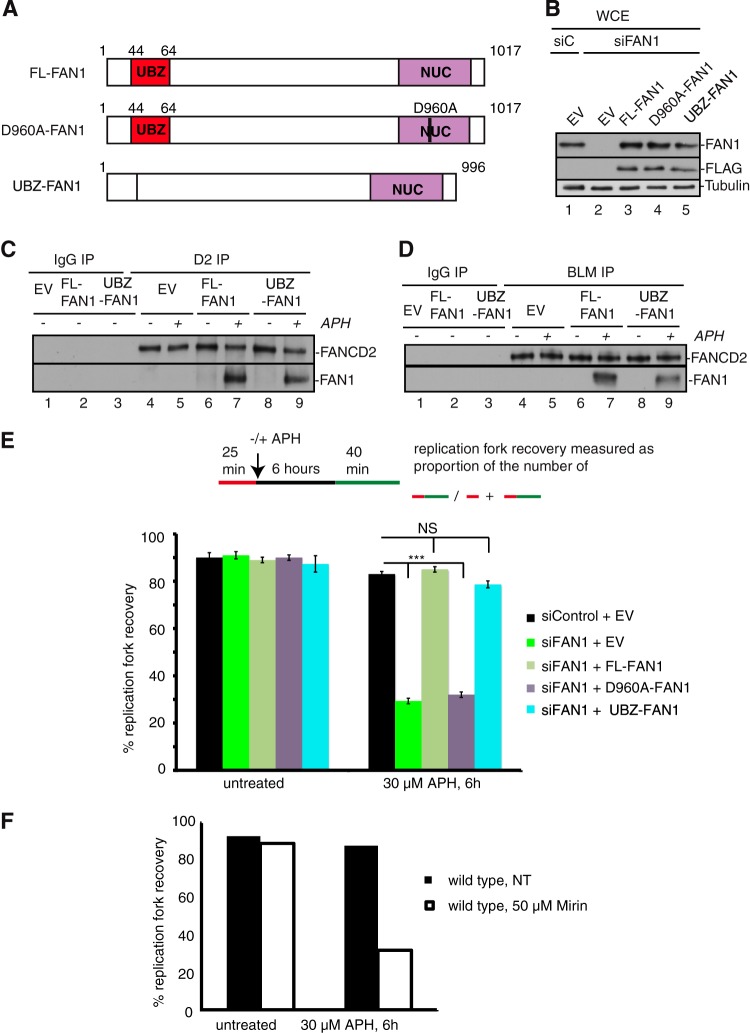
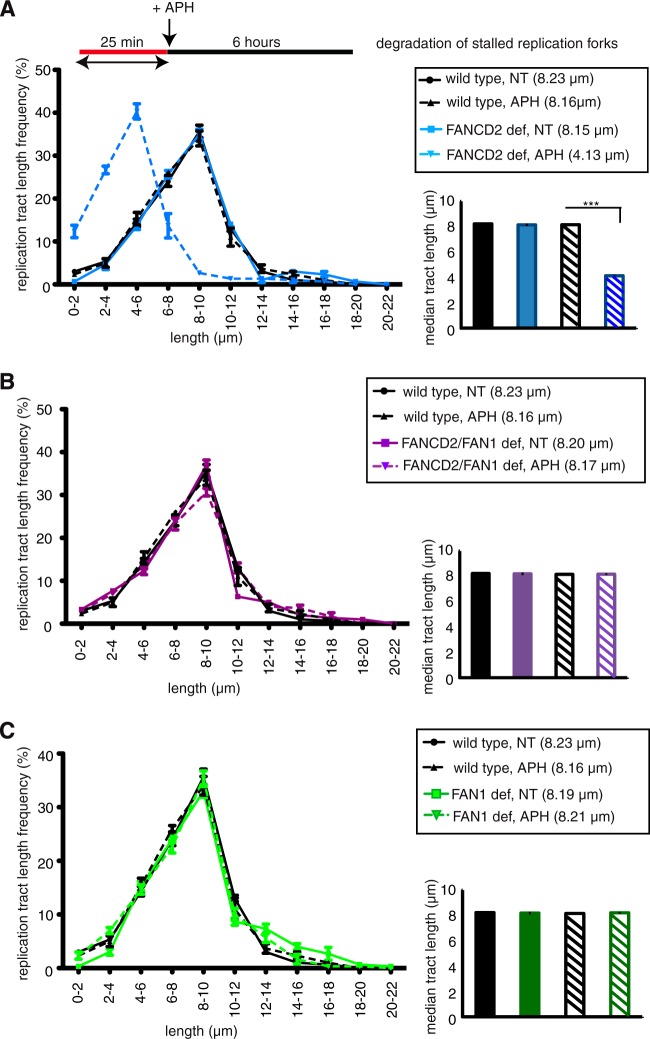
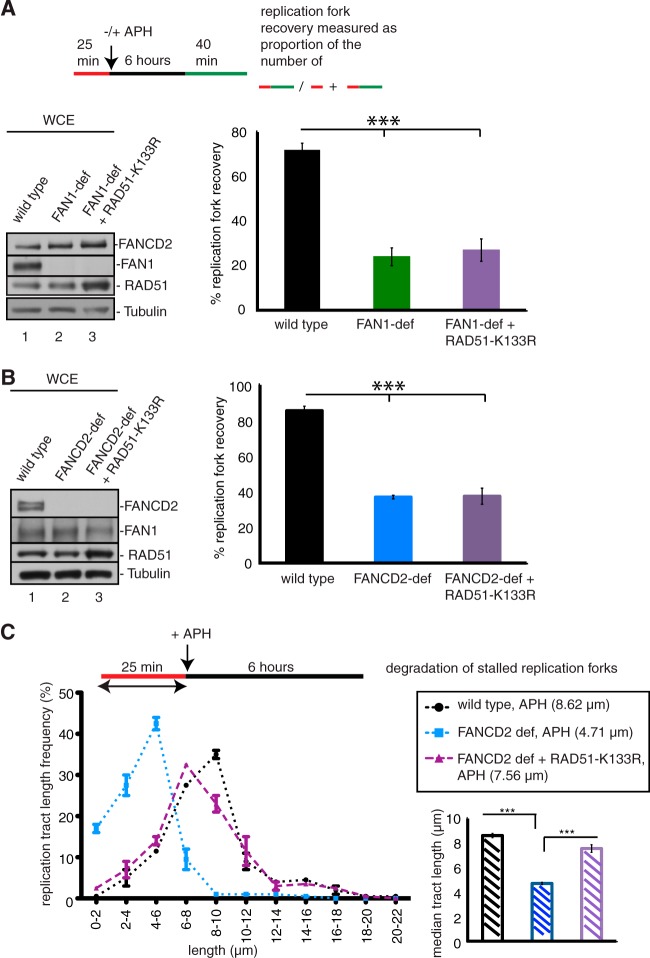
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous