

The landscape of human STR variation
- PMID: 25135957
- PMCID: PMC4216929
- DOI: 10.1101/gr.177774.114
The landscape of human STR variation
Abstract
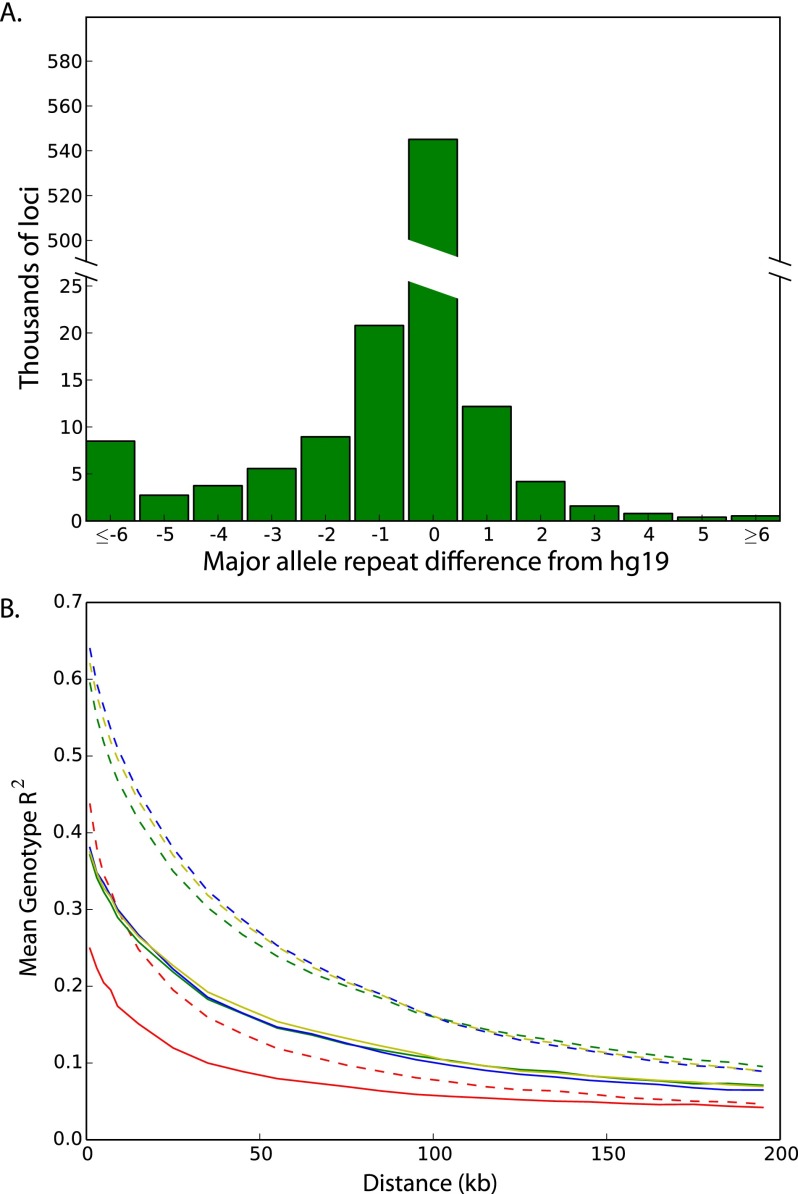
Short tandem repeats are among the most polymorphic loci in the human genome. These loci play a role in the etiology of a range of genetic diseases and have been frequently utilized in forensics, population genetics, and genetic genealogy. Despite this plethora of applications, little is known about the variation of most STRs in the human population. Here, we report the largest-scale analysis of human STR variation to date. We collected information for nearly 700,000 STR loci across more than 1000 individuals in Phase 1 of the 1000 Genomes Project. Extensive quality controls show that reliable allelic spectra can be obtained for close to 90% of the STR loci in the genome. We utilize this call set to analyze determinants of STR variation, assess the human reference genome's representation of STR alleles, find STR loci with common loss-of-function alleles, and obtain initial estimates of the linkage disequilibrium between STRs and common SNPs. Overall, these analyses further elucidate the scale of genetic variation beyond classical point mutations.
© 2014 Willems et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
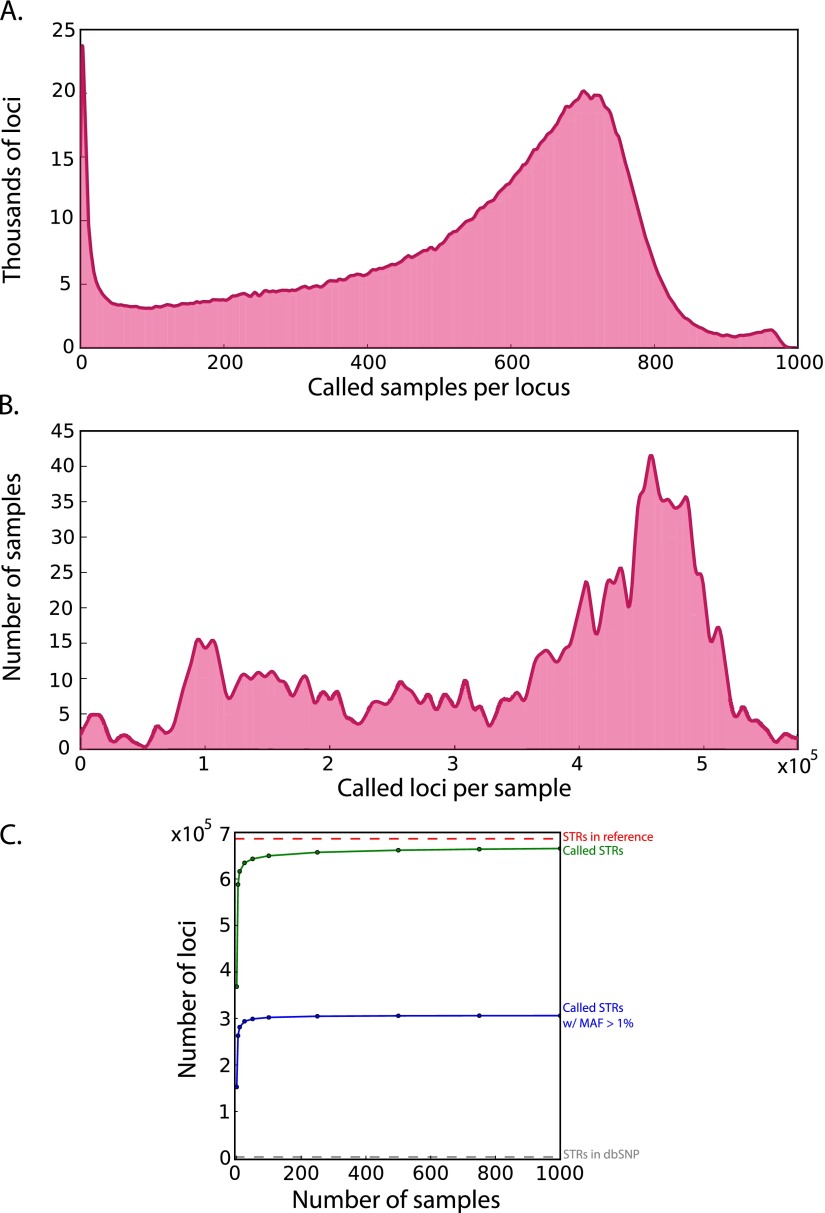
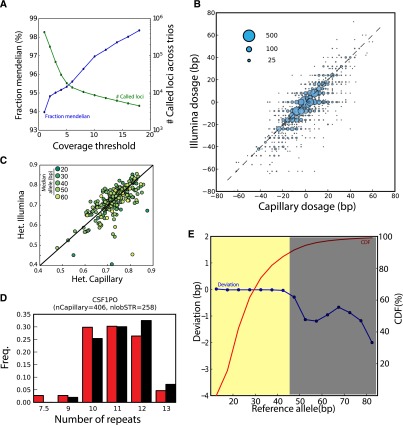
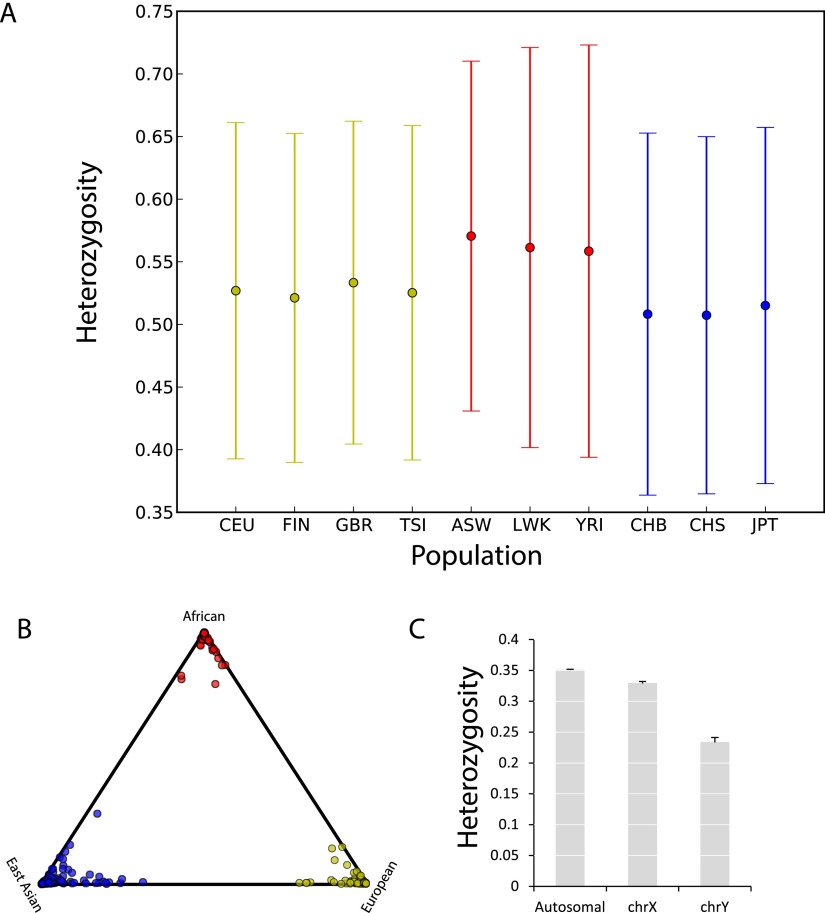
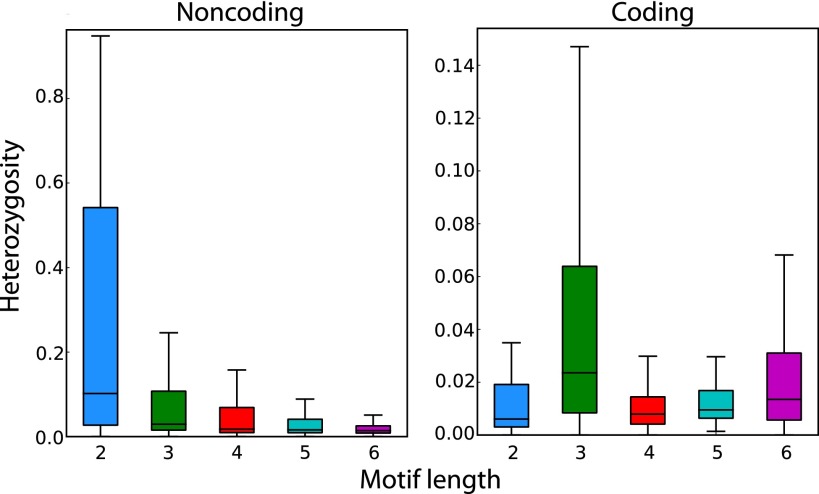
References
-
- Amiel J, Trochet D, Clément-Ziza M, Munnich A, Lyonnet S. 2004. Polyalanine expansions in human. Hum Mol Genet (suppl 2) 13: R235–R243 - PubMed
-
- Bachtrog D, Agis M, Imhof M, Schlötterer C. 2000. Microsatellite variability differs between dinucleotide repeat motifs—evidence from Drosophila melanogaster. Mol Biol Evol 17: 1277–1285 - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous