

The autosomal dominant spinocerebellar ataxias: emerging mechanistic themes suggest pervasive Purkinje cell vulnerability
- PMID: 25136055
- PMCID: PMC6718294
- DOI: 10.1136/jnnp-2014-308421
The autosomal dominant spinocerebellar ataxias: emerging mechanistic themes suggest pervasive Purkinje cell vulnerability
Abstract
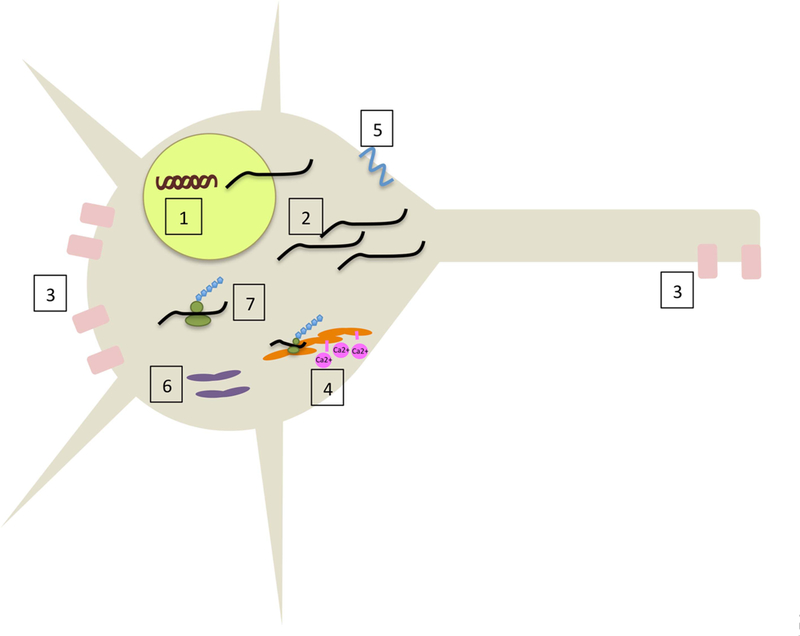
The spinocerebellar ataxias are a genetically heterogeneous group of disorders with clinically overlapping phenotypes arising from Purkinje cell degeneration, cerebellar atrophy and varying degrees of degeneration of other grey matter regions. For 22 of the 32 subtypes, a genetic cause has been identified. While recurring themes are emerging, there is no clear correlation between the clinical phenotype or penetrance, the type of genetic defect or the category of the disease mechanism, or the neuronal types involved beyond Purkinje cells. These phenomena suggest that cerebellar Purkinje cells may be a uniquely vulnerable neuronal cell type, more susceptible to a wider variety of genetic/cellular insults than most other neuron types.
Keywords: Cerebellar Ataxia; Cerebellar Degeneration; Cerebellar Disease; Genetics; Medicine.
Published by the BMJ Publishing Group Limited. For permission to use (where not already granted under a licence) please go to http://group.bmj.com/group/rights-licensing/permissions.
Figures
References
-
- Erichsen AK, Koht J, Stray-Pedersen A, et al. Prevalence of hereditary ataxia and spastic paraplegia in southeast Norway: a population-based study. Brain 2009;132:1577–88. - PubMed
-
- Coutinho P, Ruano L, Loureiro JL, et al. Hereditary ataxia and spastic paraplegia in Portugal: a population-based prevalence study. JAMA Neurol 2013;70:746–55. - PubMed
-
- Craig K, Keers SM, Archibald K, et al. Molecular epidemiology of spinocerebellar ataxia type 6. Ann Neurol 2004;55:752–5. - PubMed
-
- Hersheson J, Haworth A, Houlden H. The inherited ataxias: genetic heterogeneity, mutation databases, and future directions in research and clinical diagnostics. Hum Mutat 2012;33:1324–32. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources