

Loss of Miro1-directed mitochondrial movement results in a novel murine model for neuron disease
- PMID: 25136135
- PMCID: PMC4156725
- DOI: 10.1073/pnas.1402449111
Loss of Miro1-directed mitochondrial movement results in a novel murine model for neuron disease
Abstract
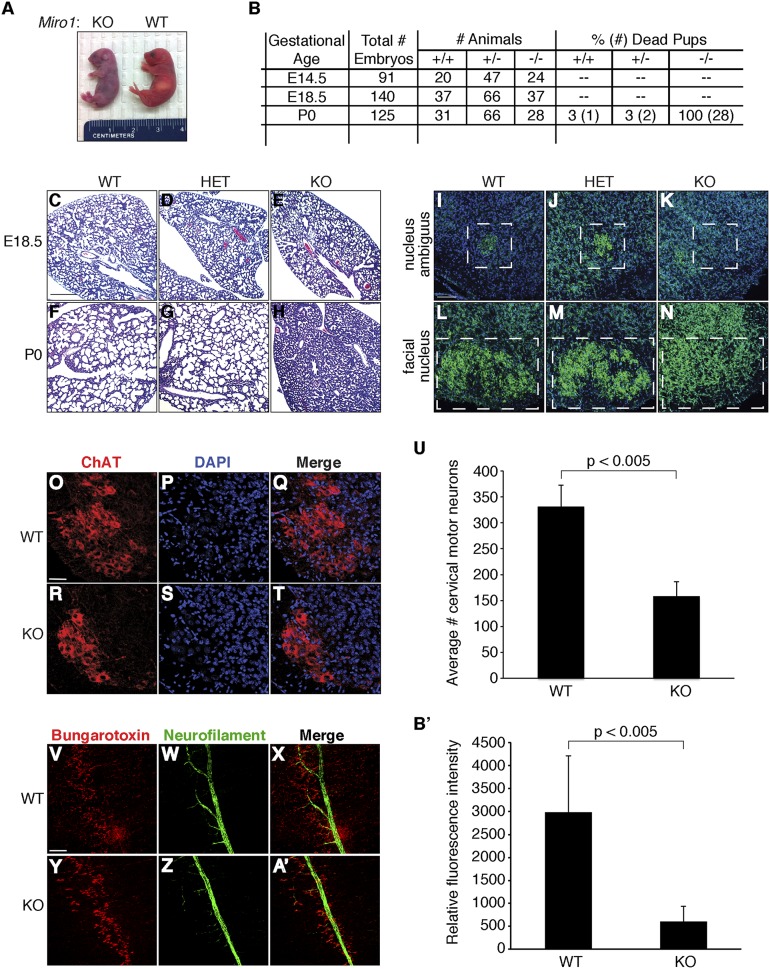
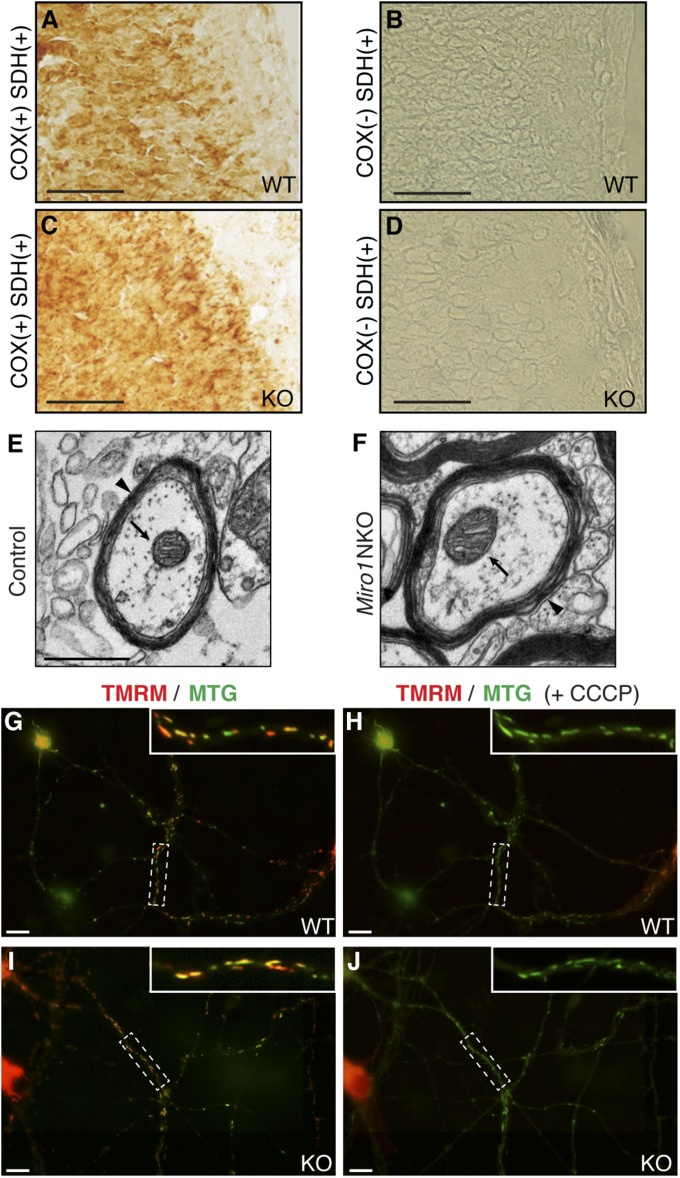
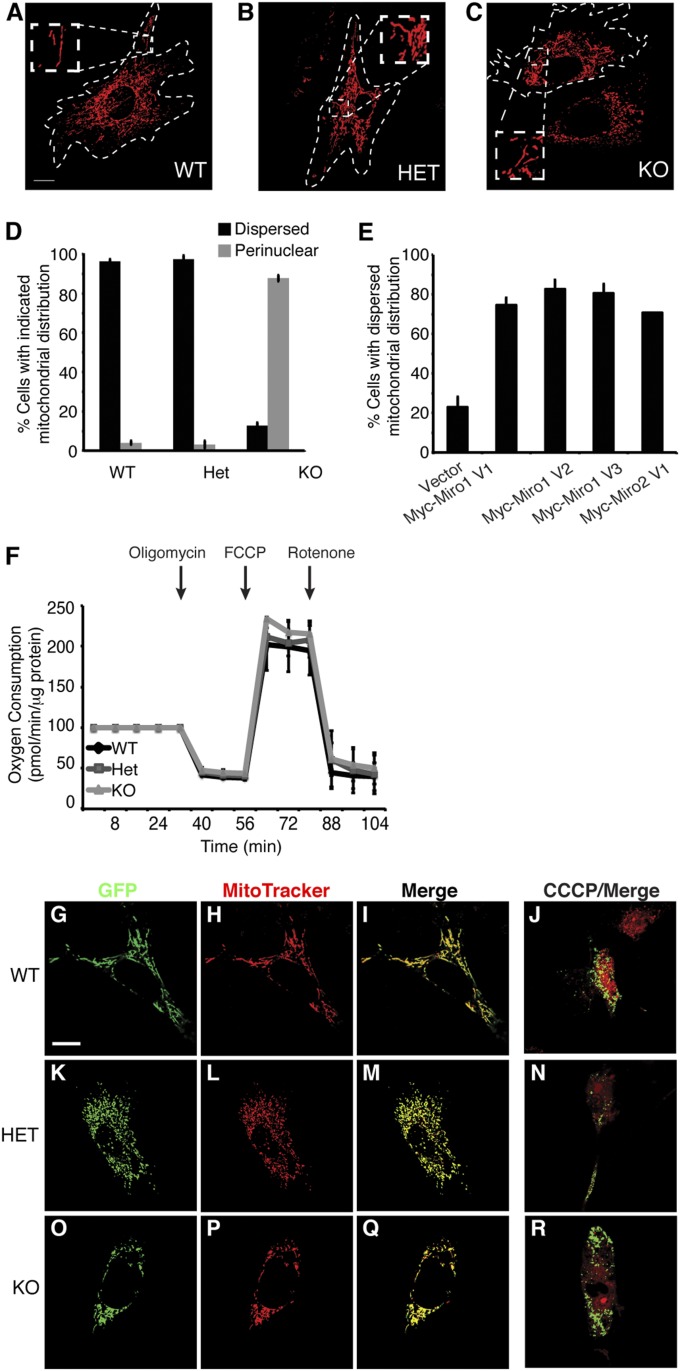
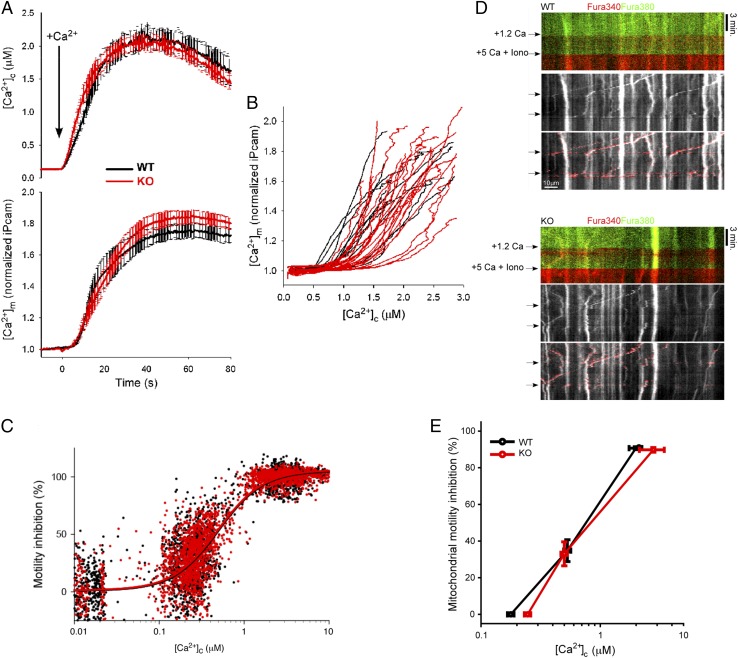
Defective mitochondrial distribution in neurons is proposed to cause ATP depletion and calcium-buffering deficiencies that compromise cell function. However, it is unclear whether aberrant mitochondrial motility and distribution alone are sufficient to cause neurological disease. Calcium-binding mitochondrial Rho (Miro) GTPases attach mitochondria to motor proteins for anterograde and retrograde transport in neurons. Using two new KO mouse models, we demonstrate that Miro1 is essential for development of cranial motor nuclei required for respiratory control and maintenance of upper motor neurons required for ambulation. Neuron-specific loss of Miro1 causes depletion of mitochondria from corticospinal tract axons and progressive neurological deficits mirroring human upper motor neuron disease. Although Miro1-deficient neurons exhibit defects in retrograde axonal mitochondrial transport, mitochondrial respiratory function continues. Moreover, Miro1 is not essential for calcium-mediated inhibition of mitochondrial movement or mitochondrial calcium buffering. Our findings indicate that defects in mitochondrial motility and distribution are sufficient to cause neurological disease.
Keywords: Ca2+-dependent motility; Miro GTPase; mitochondrial respiration.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
