

Wood smoke enhances cigarette smoke-induced inflammation by inducing the aryl hydrocarbon receptor repressor in airway epithelial cells
- PMID: 25137396
- PMCID: PMC4370262
- DOI: 10.1165/rcmb.2014-0142OC
Wood smoke enhances cigarette smoke-induced inflammation by inducing the aryl hydrocarbon receptor repressor in airway epithelial cells
Abstract
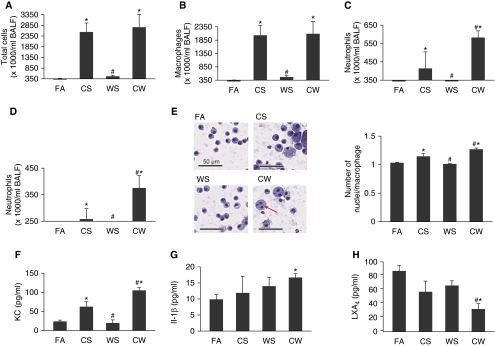
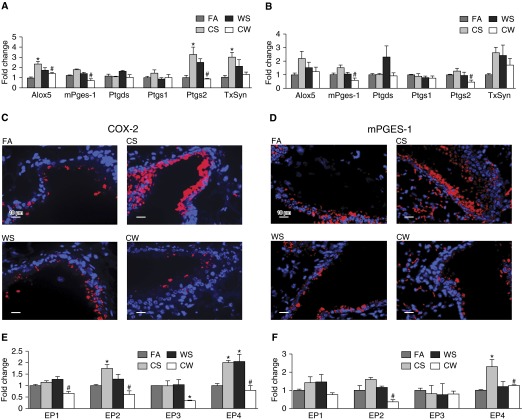
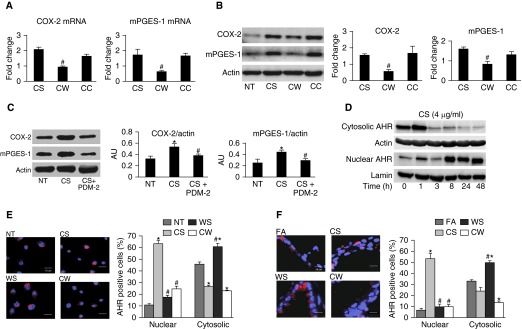
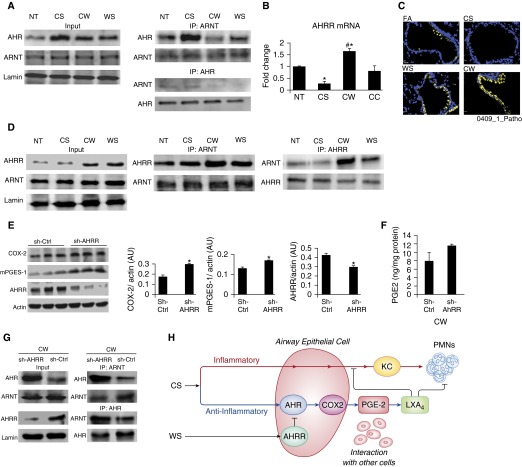
Our previous studies showed that cigarette smokers who are exposed to wood smoke (WS) are at an increased risk for chronic bronchitis and reduced lung function. The present study was undertaken to determine the mechanisms for WS-induced adverse effects. We studied the effect of WS exposure using four cohorts of mice. C57Bl/6 mice were exposed for 4 or 12 weeks to filtered air, to 10 mg/m(3) WS for 2 h/d, to 250 mg/m(3) cigarette smoke (CS) for 6 h/d, or to CS followed by WS (CW). Inflammation was absent in the filtered air and WS groups, but enhanced by twofold in the bronchoalveolar lavage of the CW compared with CS group as measured by neutrophil numbers and levels of the neutrophil chemoattractant, keratinocyte-derived chemokine. The levels of the anti-inflammatory lipoxin, lipoxin A4, were reduced by threefold along with cyclo-oxygenase (COX)-2 and microsomal prostaglandin E synthase (mPGES)-1 in airway epithelial cells and PGE2 levels in the bronchoalveolar lavage of CW compared with CS mice. We replicated, in primary human airway epithelial cells, the changes observed in mice. Immunoprecipitations showed that WS blocked the interaction of aryl hydrocarbon receptor (AHR) with AHR nuclear transporter to reduce expression of COX-2 and mPGES-1 by increasing expression of AHR repressor (AHRR). Collectively, these studies show that exposure to low concentrations of WS enhanced CS-induced inflammation by inducing AHRR expression to suppress AHR, COX-2, and mPGES-1 expression, and levels of PGE2 and lipoxin A4. Therefore, AHRR is a potential therapeutic target for WS-associated exacerbations of CS-induced inflammation.
Keywords: air pollution; arachidonic acid pathway; exacerbation; lipoxin; neutrophilic inflammation.
Figures
References
-
- Larson TV, Koenig JQ. Wood smoke: emissions and noncancer respiratory effects. Annu Rev Public Health. 1994;15:133–156. - PubMed
-
- Orozco-Levi M, Garcia-Aymerich J, Villar J, Ramírez-Sarmiento A, Antó JM, Gea J. Wood smoke exposure and risk of chronic obstructive pulmonary disease. Eur Respir J. 2006;27:542–546. - PubMed
-
- Gan WQ, FitzGerald JM, Carlsten C, Sadatsafavi M, Brauer M. Associations of ambient air pollution with chronic obstructive pulmonary disease hospitalization and mortality. Am J Respir Crit Care Med. 2013;187:721–727. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
