

Aberrant regulation and function of microRNAs in cancer
- PMID: 25137592
- PMCID: PMC4177046
- DOI: 10.1016/j.cub.2014.06.043
Aberrant regulation and function of microRNAs in cancer
Abstract
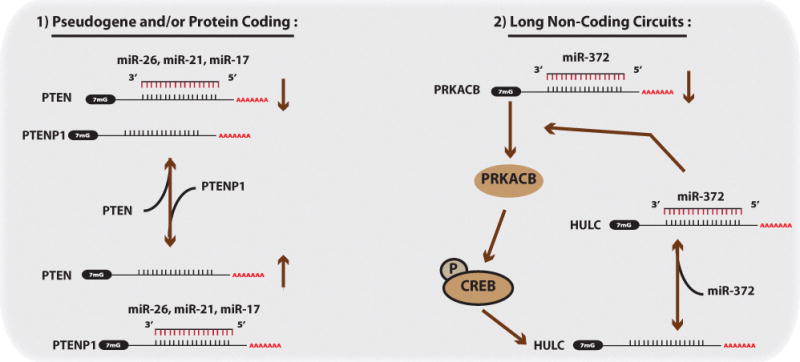
Malignant neoplasms are consistently among the top four leading causes of death in all age groups in the United States, despite a concerted effort toward developing novel therapeutic approaches. Our understanding of and therapeutic strategy for treating each of these neoplastic diseases have been improved through decades of research on the genetics, signaling pathways, and cellular biology that govern tumor cell initiation, progression and maintenance. Much of this work has concentrated on post-translational modifications and abnormalities at the DNA level, including point mutations, amplifications/deletions, and chromosomal translocations, and how these aberrant events affect the expression and function of protein-coding genes. Only recently has a novel class of conserved gene regulatory molecules been identified as a major contributor to malignant neoplastic disease. This review focuses on how these small non-coding RNA molecules, termed microRNAs (miRNAs), can function as oncogenes or tumor suppressors, and how the misexpression of miRNAs and dysregulation of factors that regulate miRNAs contribute to the tumorigenic process. Specific focus is given to more recently discovered regulatory mechanisms that go awry in cancer, and how these changes alter miRNA expression, processing, and function.
Copyright © 2014 Elsevier Ltd. All rights reserved.
Conflict of interest statement
The authors have no conflicts of interest to disclose.
Figures
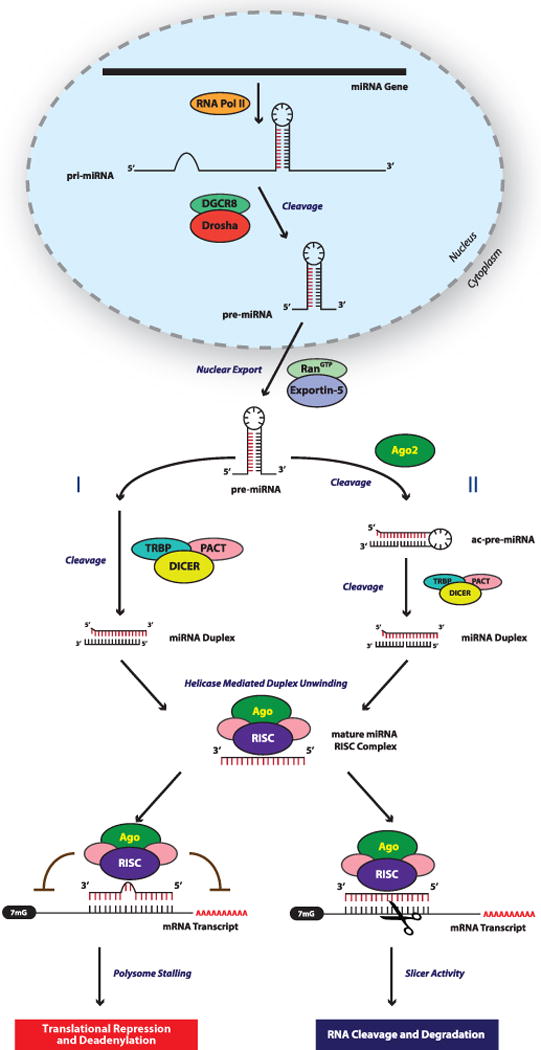
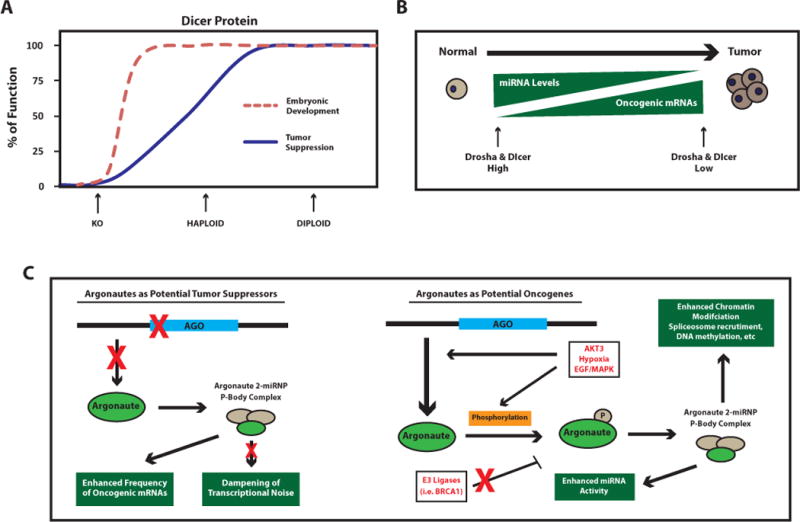

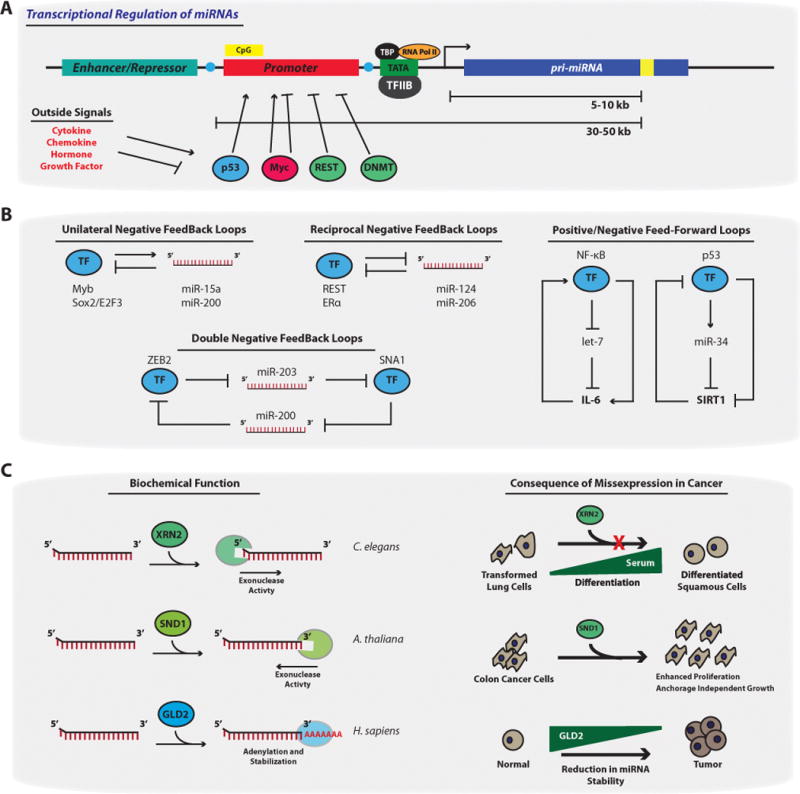
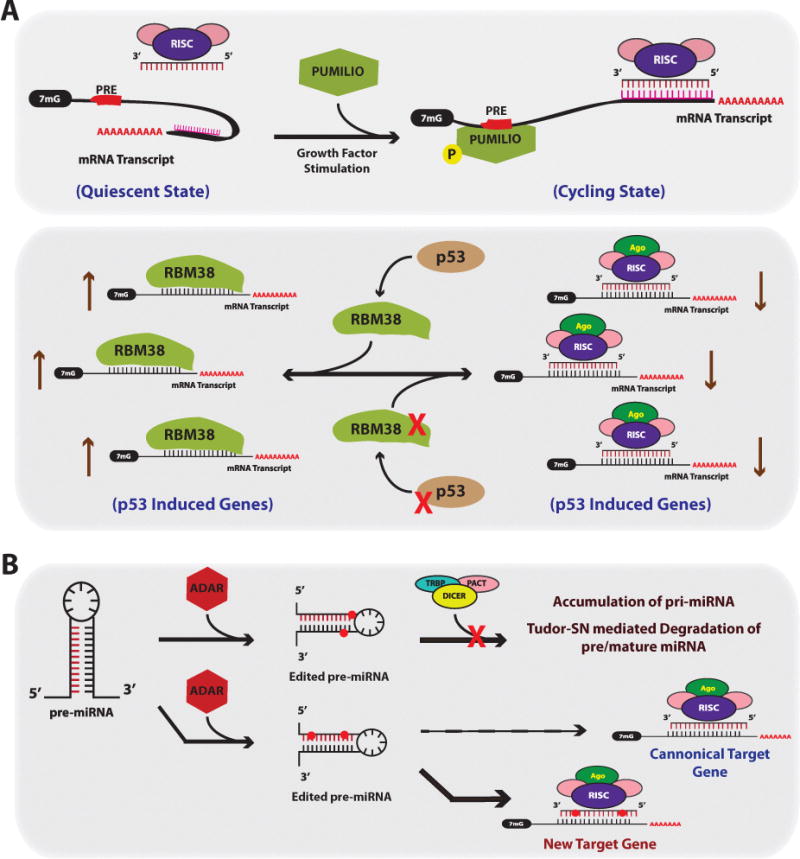
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
