

Ubiquitin-binding site 2 of ataxin-3 prevents its proteasomal degradation by interacting with Rad23
- PMID: 25144244
- PMCID: PMC4237202
- DOI: 10.1038/ncomms5638
Ubiquitin-binding site 2 of ataxin-3 prevents its proteasomal degradation by interacting with Rad23
Abstract
Polyglutamine repeat expansion in ataxin-3 causes neurodegeneration in the most common dominant ataxia, spinocerebellar ataxia type 3 (SCA3). Since reducing levels of disease proteins improves pathology in animals, we investigated how ataxin-3 is degraded. Here we show that, unlike most proteins, ataxin-3 turnover does not require its ubiquitination, but is regulated by ubiquitin-binding site 2 (UbS2) on its N terminus. Mutating UbS2 decreases ataxin-3 protein levels in cultured mammalian cells and in Drosophila melanogaster by increasing its proteasomal turnover. Ataxin-3 interacts with the proteasome-associated proteins Rad23A/B through UbS2. Knockdown of Rad23 in cultured cells and in Drosophila results in lower levels of ataxin-3 protein. Importantly, reducing Rad23 suppresses ataxin-3-dependent degeneration in flies. We present a mechanism for ubiquitination-independent degradation that is impeded by protein interactions with proteasome-associated factors. We conclude that UbS2 is a potential target through which to enhance ataxin-3 degradation for SCA3 therapy.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
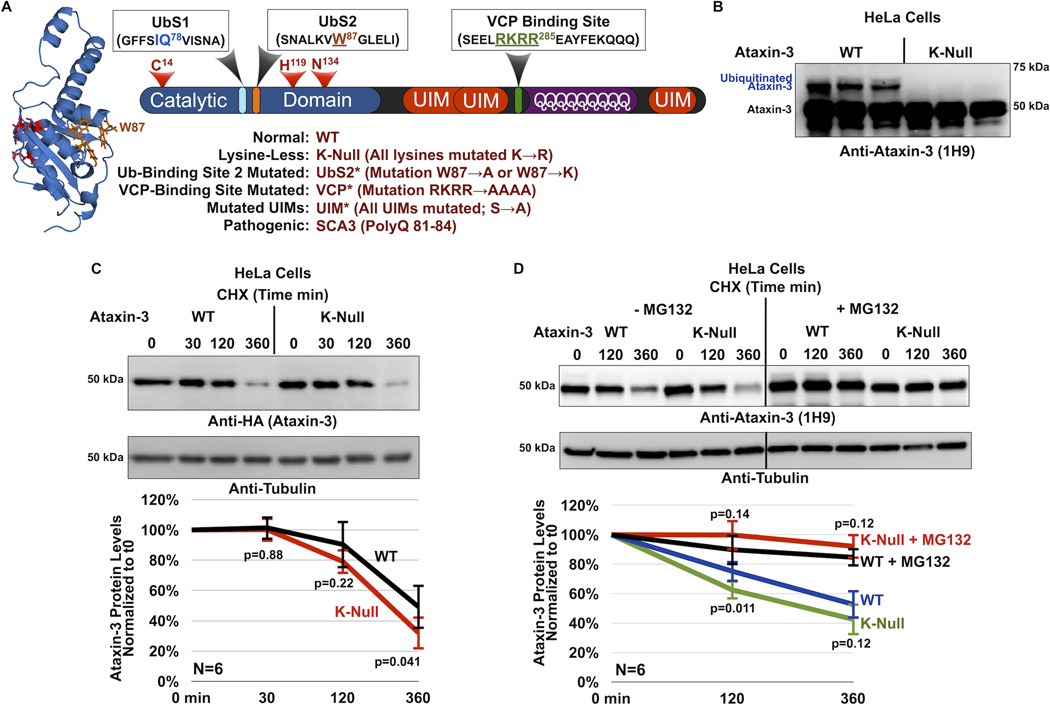
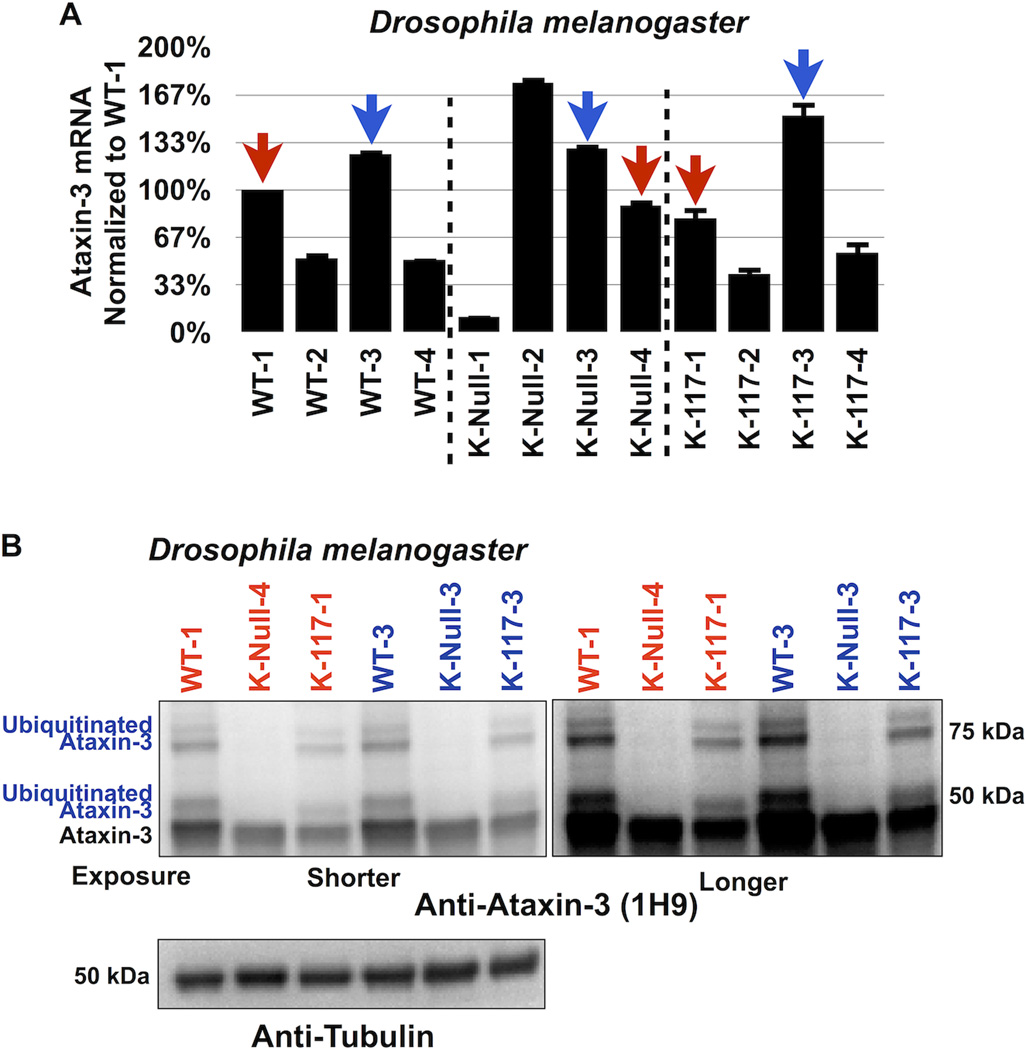
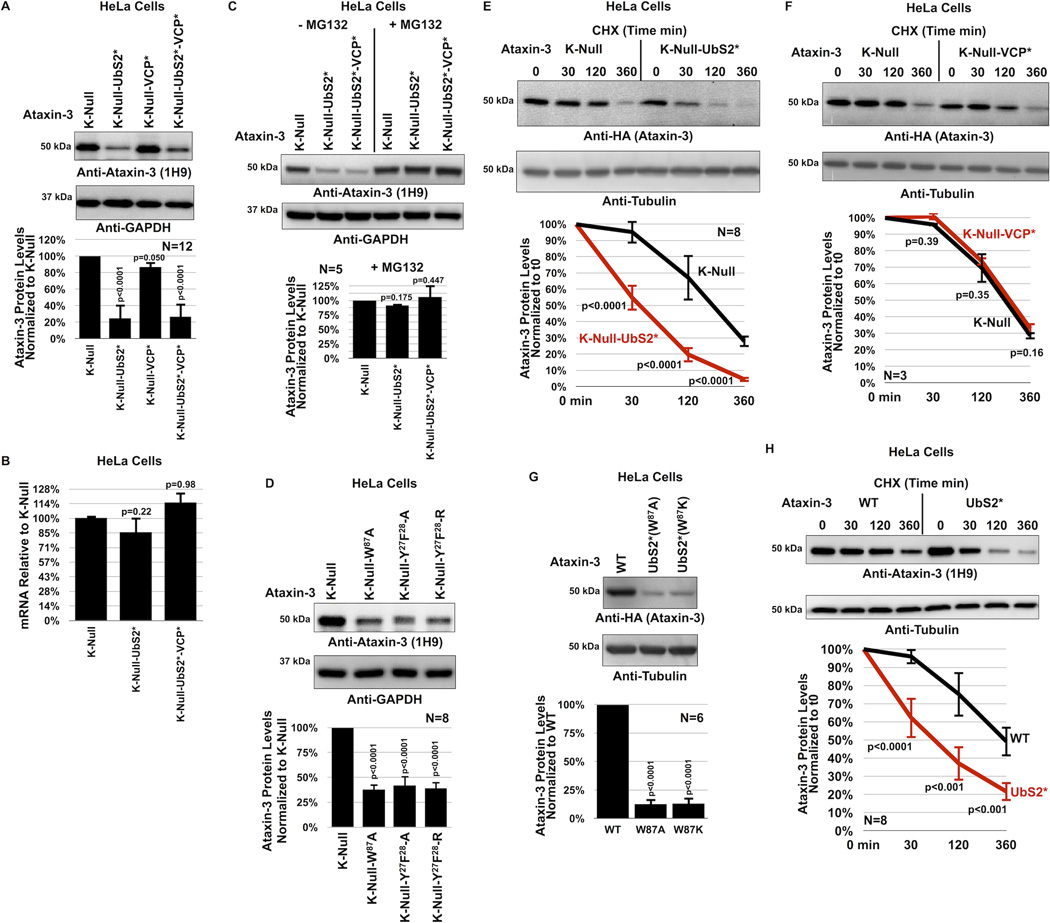
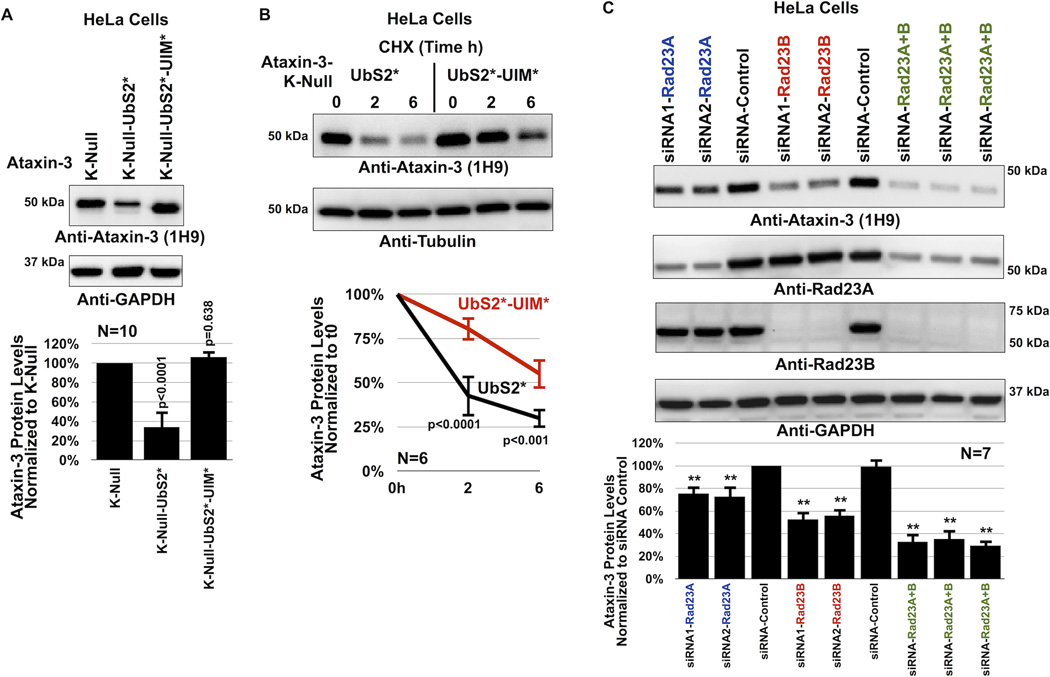
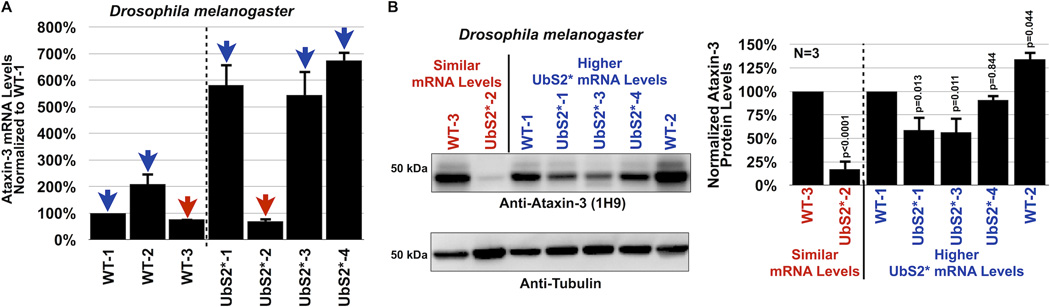
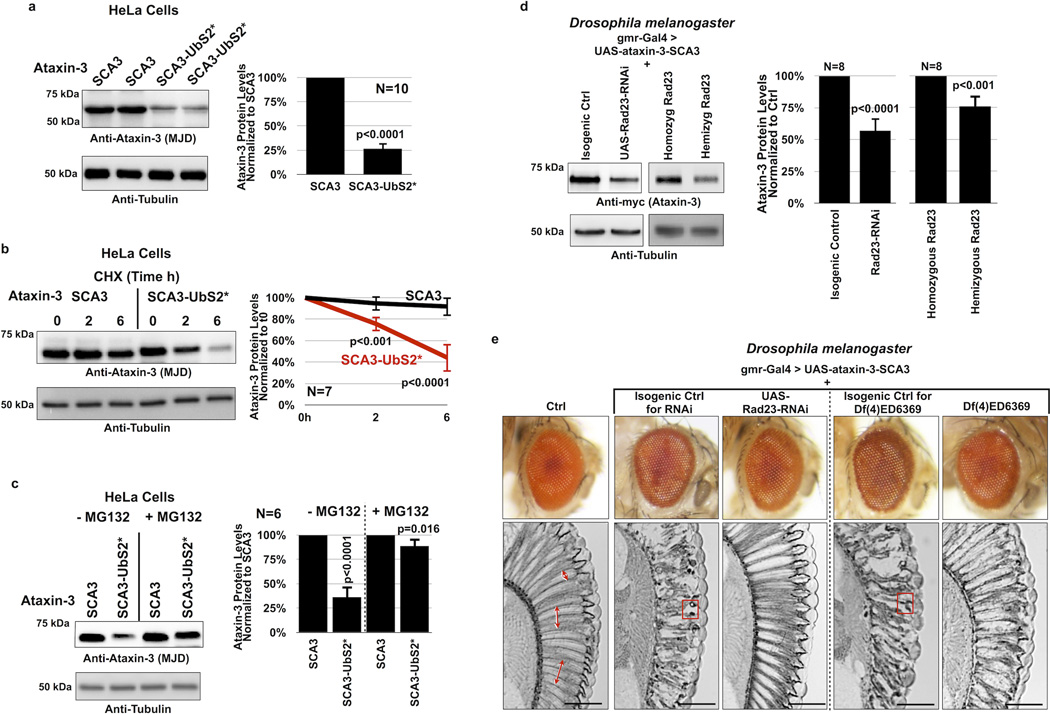
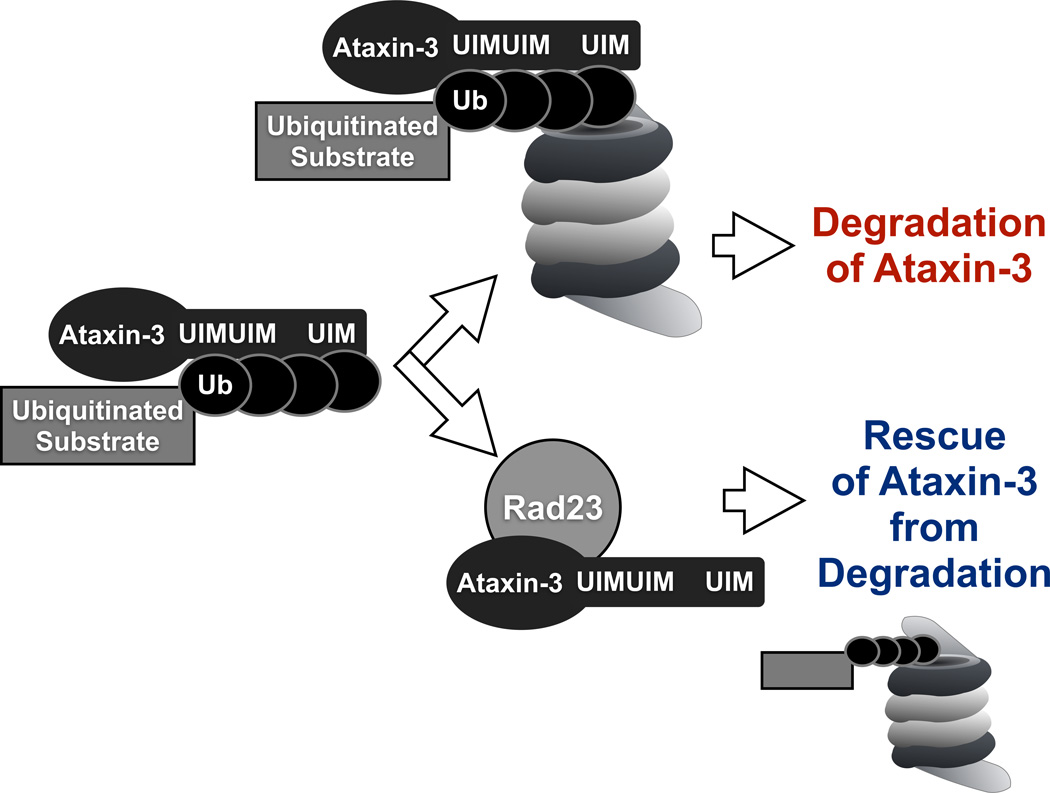
References
-
- Todi SV, Williams A, Paulson HL. Polyglutamine Repeat Disorders, including Huntington’s Disease. In: Waxman SG, editor. Molecular Neurology. 1st edition. Academic Press; 2007.
-
- Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Ann. Rev. Neurosci. 2007;30:575–621. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
