

Genetic and laboratory diagnostic approach in Niemann Pick disease type C
- PMID: 25145893
- PMCID: PMC4141153
- DOI: 10.1007/s00415-014-7386-8
Genetic and laboratory diagnostic approach in Niemann Pick disease type C
Abstract
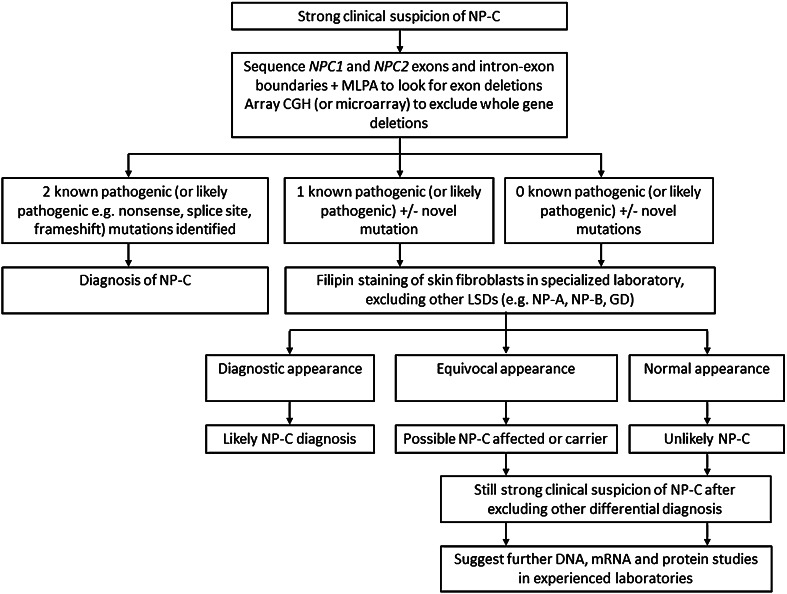
Niemann Pick disease type C (NP-C) is a rare autosomal recessive disorder that results from mutations in either the NPC1 or the NPC2 gene. The estimated incidence of NP-C is 1 in 120,000 live births, although the frequency of cases is higher in some isolated populations. More than 350 different NPC1 and NPC2 gene mutations have been reported in patients with confirmed diagnoses. Approximately 95 % of patients harbour mutations in NPC1, with most of the remaining patients having NPC2 mutations. The traditional methods for diagnosing patients with NP-C include histopathological analysis of bone marrow aspirate, liver and skin biopsies, fluorescent and electron microscopy, and cholesterol esterification assays. New laboratory methods that use mass spectroscopy for detection of cholesterol metabolism products are promising to become part of the routine diagnostic and screening tests in the near future, but further evaluation is required to determine the sensitivity and specificity of these analyses in patients with different age-at-onset forms of NP-C. Although filipin staining and cholesterol esterification studies performed in patient skin fibroblasts can, in experienced hands, provide a robust approach to diagnosing NP-C, they are only available in a few specialist laboratories. Thus, sequencing of NPC1 and NPC2 is currently the most universally accessible diagnostic technique in this disorder.
Figures
References
-
- Alvelius G, Hjalmarson O, Griffiths WJ, Bjorkhem I, Sjovall J. Identification of unusual 7-oxygenated bile acid sulfates in a patient with Niemann–Pick disease, type C. J Lipid Res. 2001;42:1571–1577. - PubMed
-
- Argoff CE, Comly ME, Blanchette-Mackie J, Kruth HS, Pye HT, Goldin E, Kaneski C, Vanier MT, et al. Type C Niemann–Pick disease: cellular uncoupling of cholesterol homeostasis is linked to the severity of disruption in the intracellular transport of exogenously derived cholesterol. Biochim Biophys Acta. 1991;1096:319–327. doi: 10.1016/0925-4439(91)90068-K. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
