

Urinary tract effects of HPSE2 mutations
- PMID: 25145936
- PMCID: PMC4378092
- DOI: 10.1681/ASN.2013090961
Urinary tract effects of HPSE2 mutations
Abstract
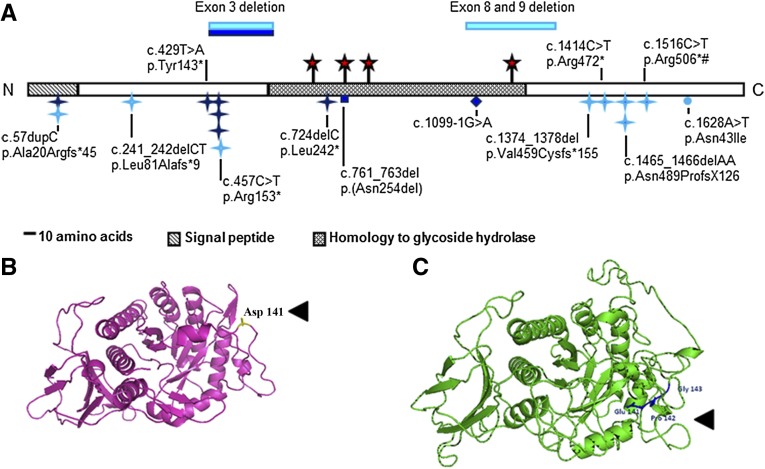
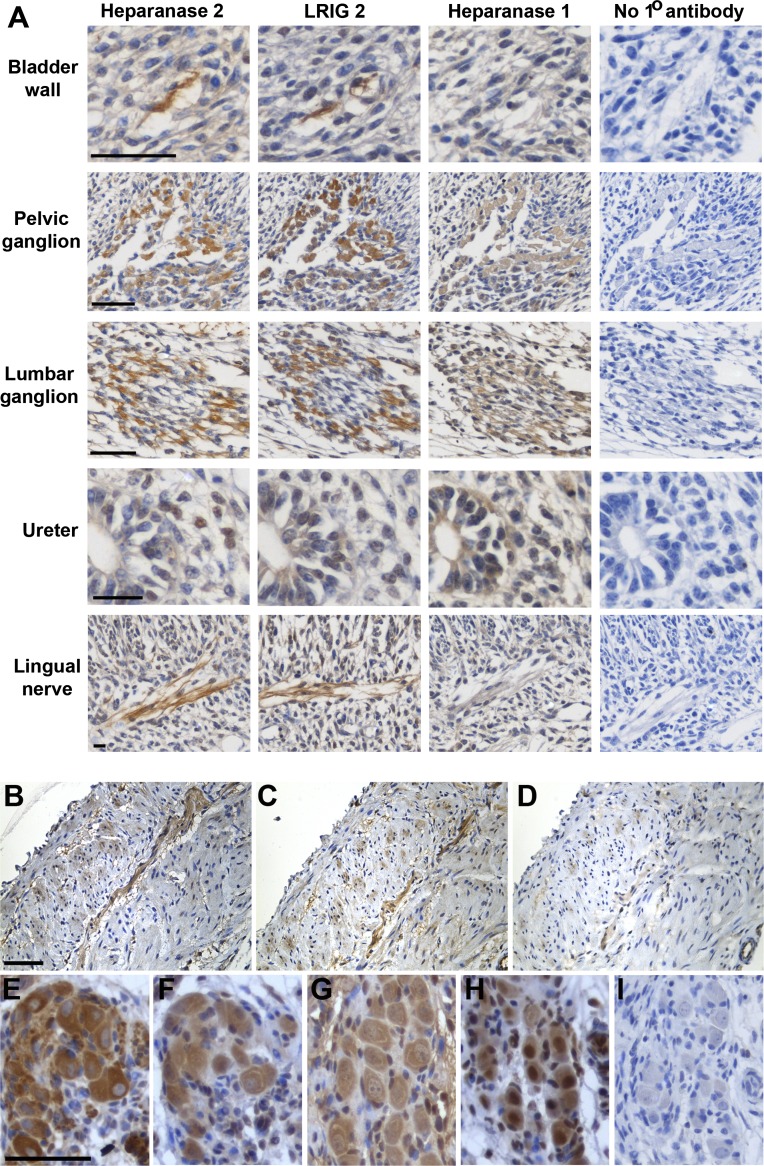
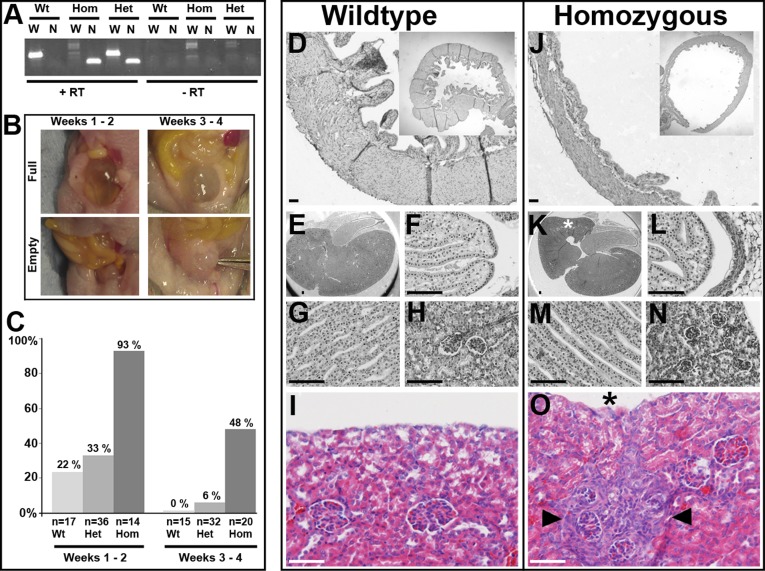
Urofacial syndrome (UFS) is an autosomal recessive congenital disease featuring grimacing and incomplete bladder emptying. Mutations of HPSE2, encoding heparanase 2, a heparanase 1 inhibitor, occur in UFS, but knowledge about the HPSE2 mutation spectrum is limited. Here, seven UFS kindreds with HPSE2 mutations are presented, including one with deleted asparagine 254, suggesting a role for this amino acid, which is conserved in vertebrate orthologs. HPSE2 mutations were absent in 23 non-neurogenic neurogenic bladder probands and, of 439 families with nonsyndromic vesicoureteric reflux, only one carried a putative pathogenic HPSE2 variant. Homozygous Hpse2 mutant mouse bladders contained urine more often than did wild-type organs, phenocopying human UFS. Pelvic ganglia neural cell bodies contained heparanase 1, heparanase 2, and leucine-rich repeats and immunoglobulin-like domains-2 (LRIG2), which is mutated in certain UFS families. In conclusion, heparanase 2 is an autonomic neural protein implicated in bladder emptying, but HPSE2 variants are uncommon in urinary diseases resembling UFS.
Keywords: genetics and development; human genetics; molecular genetics; pediatric nephrology.
Copyright © 2015 by the American Society of Nephrology.
Figures
References
-
- Online Mendelian Inheritance in Man. Available at: http://www.ncbi.nlm.nih.gov/omim. Accessed April 25, 2014
-
- Ochoa B: Can a congenital dysfunctional bladder be diagnosed from a smile? The Ochoa syndrome updated. Pediatr Nephrol 19: 6–12, 2004 - PubMed
-
- Newman WG, Woolf AS, Stuart HM: Urofacial syndrome. GeneReviews 1993–2013, 2013. Available at: http://www.ncbi.nlm.nih.gov/books/NBK154138/. Accessed April 24, 2014
-
- Daly SB, Urquhart JE, Hilton E, McKenzie EA, Kammerer RA, Lewis M, Kerr B, Stuart H, Donnai D, Long DA, Burgu B, Aydogdu O, Derbent M, Garcia-Minaur S, Reardon W, Gener B, Shalev S, Smith R, Woolf AS, Black GC, Newman WG: Mutations in HPSE2 cause urofacial syndrome. Am J Hum Genet 86: 963–969, 2010 - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
