

Assays for the identification and prioritization of drug candidates for spinal muscular atrophy
- PMID: 25147906
- PMCID: PMC4142828
- DOI: 10.1089/adt.2014.587
Assays for the identification and prioritization of drug candidates for spinal muscular atrophy
Abstract
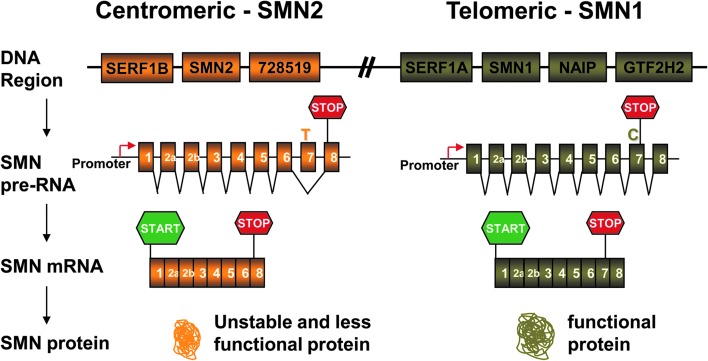
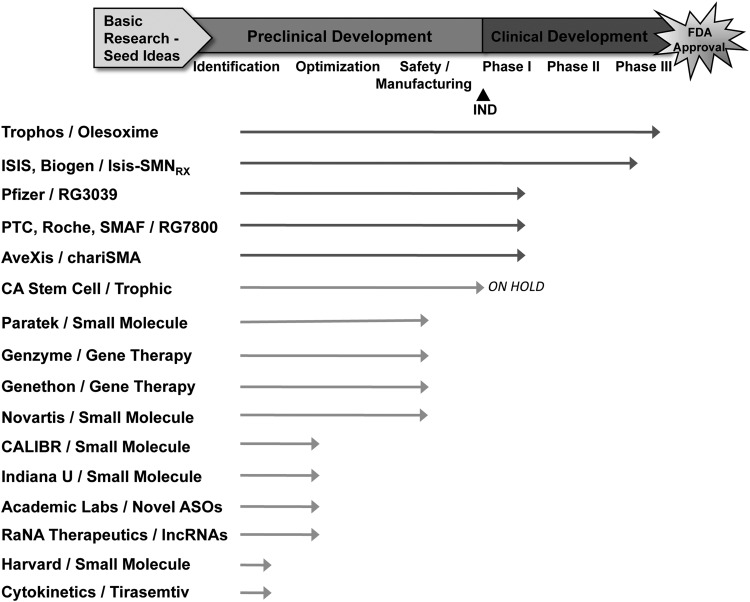
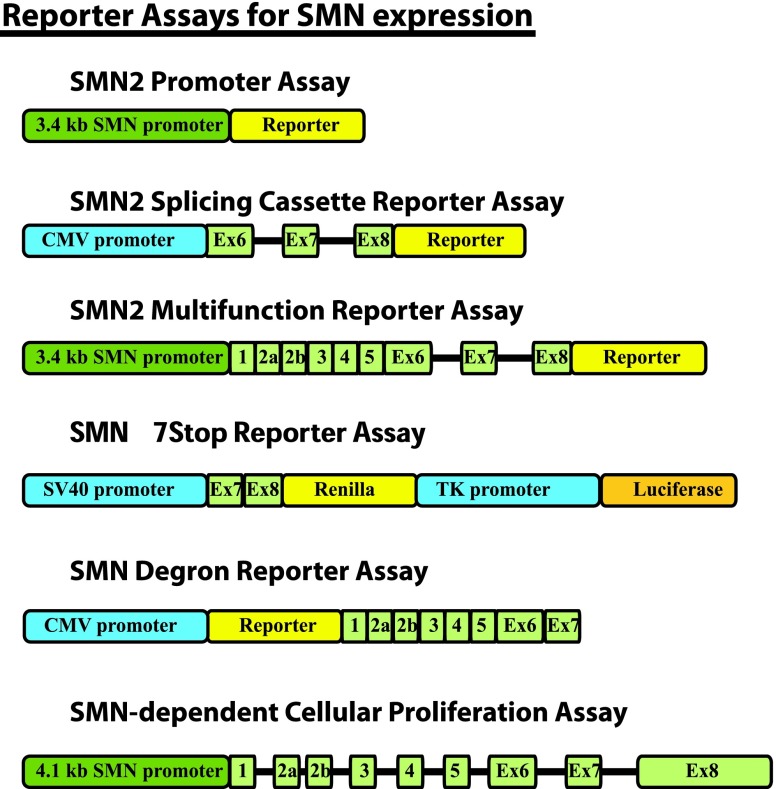
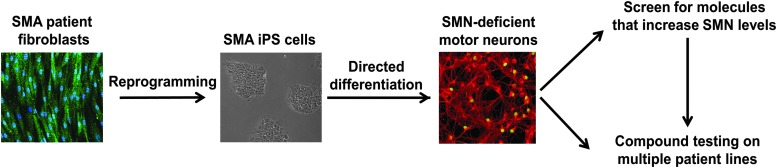
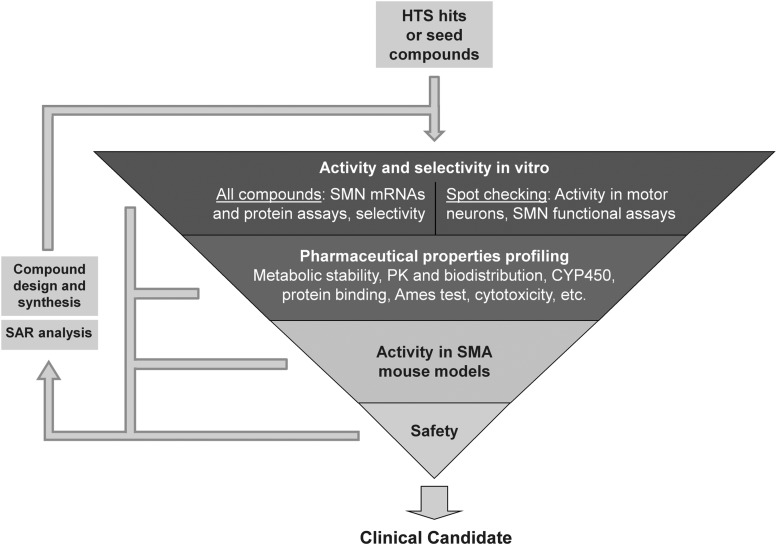
Spinal muscular atrophy (SMA) is an autosomal recessive genetic disorder resulting in degeneration of α-motor neurons of the anterior horn and proximal muscle weakness. It is the leading cause of genetic mortality in children younger than 2 years. It affects ∼1 in 11,000 live births. In 95% of cases, SMA is caused by homozygous deletion of the SMN1 gene. In addition, all patients possess at least one copy of an almost identical gene called SMN2. A single point mutation in exon 7 of the SMN2 gene results in the production of low levels of full-length survival of motor neuron (SMN) protein at amounts insufficient to compensate for the loss of the SMN1 gene. Although no drug treatments are available for SMA, a number of drug discovery and development programs are ongoing, with several currently in clinical trials. This review describes the assays used to identify candidate drugs for SMA that modulate SMN2 gene expression by various means. Specifically, it discusses the use of high-throughput screening to identify candidate molecules from primary screens, as well as the technical aspects of a number of widely used secondary assays to assess SMN messenger ribonucleic acid (mRNA) and protein expression, localization, and function. Finally, it describes the process of iterative drug optimization utilized during preclinical SMA drug development to identify clinical candidates for testing in human clinical trials.
Figures
References
-
- Crawford TO: From enigmatic to problematic: the new molecular genetics of childhood spinal muscular atrophy. Neurology 1996;46:335–340 - PubMed
-
- Pearn J: Autosomal dominant spinal muscular atrophy: a clinical and genetic study. J Neurol Sci 1978;38:263–275 - PubMed
-
- Wang CH, Finkel RS, Bertini ES, et al. : Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol 2007;22:1027–1049 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
