

Multi-platform assessment of transcriptome profiling using RNA-seq in the ABRF next-generation sequencing study
- PMID: 25150835
- PMCID: PMC4167418
- DOI: 10.1038/nbt.2972
Multi-platform assessment of transcriptome profiling using RNA-seq in the ABRF next-generation sequencing study
Erratum in
- Nat Biotechnol. 2014 Nov;32(11):1166. Rosenfeld, Jeffrey [corrected to Rosenfeld, Jeffrey A]
Abstract
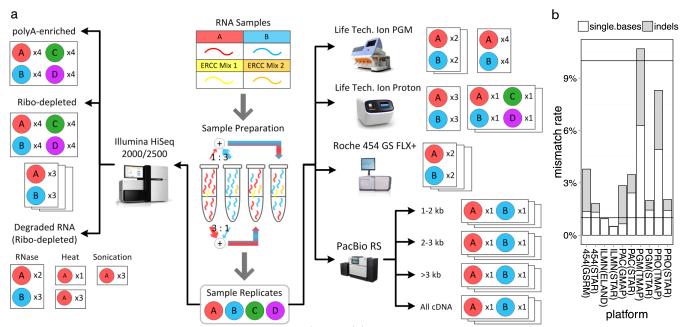
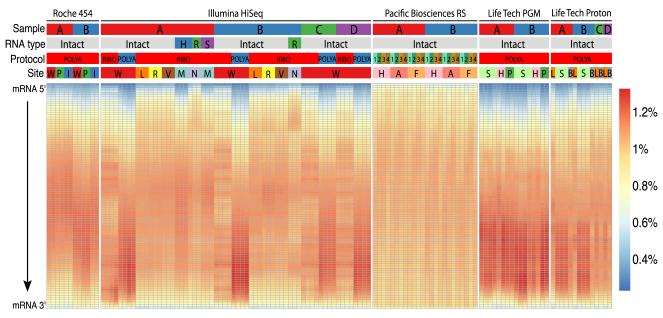
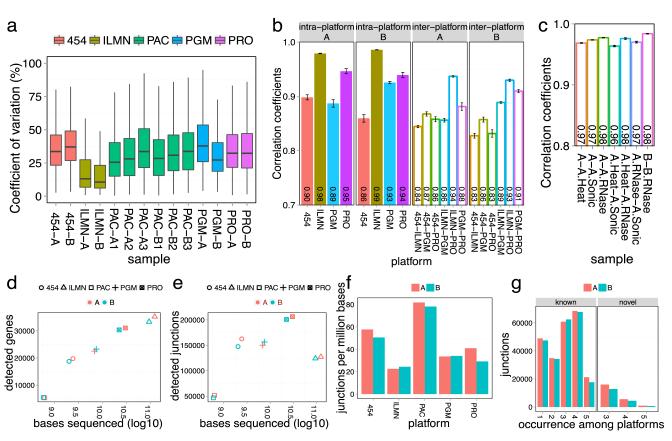
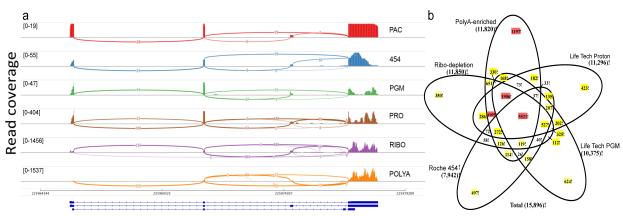
High-throughput RNA sequencing (RNA-seq) greatly expands the potential for genomics discoveries, but the wide variety of platforms, protocols and performance capabilitites has created the need for comprehensive reference data. Here we describe the Association of Biomolecular Resource Facilities next-generation sequencing (ABRF-NGS) study on RNA-seq. We carried out replicate experiments across 15 laboratory sites using reference RNA standards to test four protocols (poly-A-selected, ribo-depleted, size-selected and degraded) on five sequencing platforms (Illumina HiSeq, Life Technologies PGM and Proton, Pacific Biosciences RS and Roche 454). The results show high intraplatform (Spearman rank R > 0.86) and inter-platform (R > 0.83) concordance for expression measures across the deep-count platforms, but highly variable efficiency and cost for splice junction and variant detection between all platforms. For intact RNA, gene expression profiles from rRNA-depletion and poly-A enrichment are similar. In addition, rRNA depletion enables effective analysis of degraded RNA samples. This study provides a broad foundation for cross-platform standardization, evaluation and improvement of RNA-seq.
Figures
References
-
- Nagalakshmi U, Waern K, Snyder M. RNA-Seq: a method for comprehensive transcriptome analysis. Curr Protoc Mol Biol. 2010;11:11–13. Chapter 4, Unit 4. - PubMed
-
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. - PubMed
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
