

Normalization of RNA-seq data using factor analysis of control genes or samples
- PMID: 25150836
- PMCID: PMC4404308
- DOI: 10.1038/nbt.2931
Normalization of RNA-seq data using factor analysis of control genes or samples
Abstract
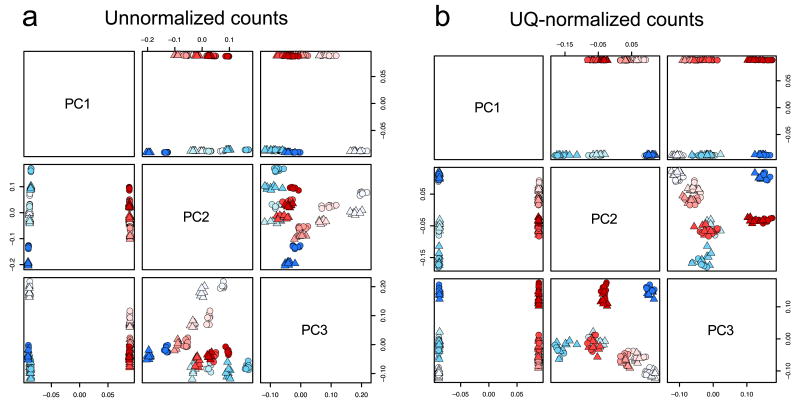
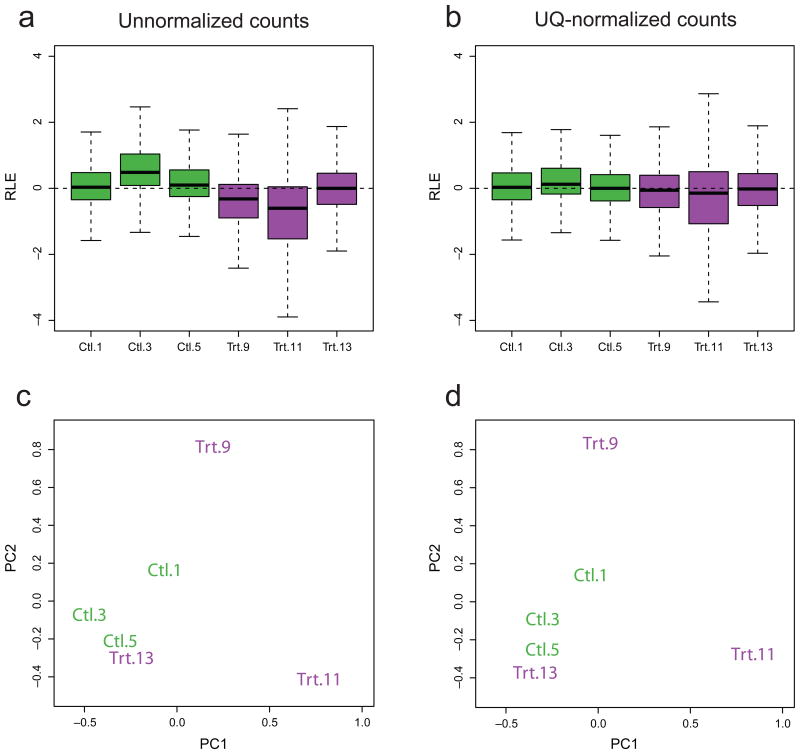
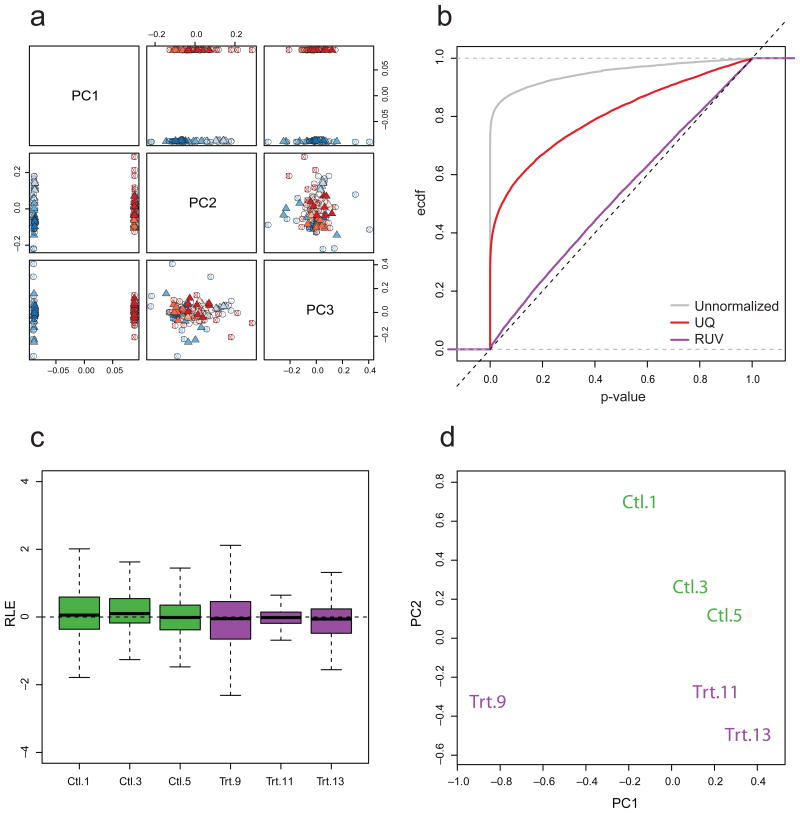
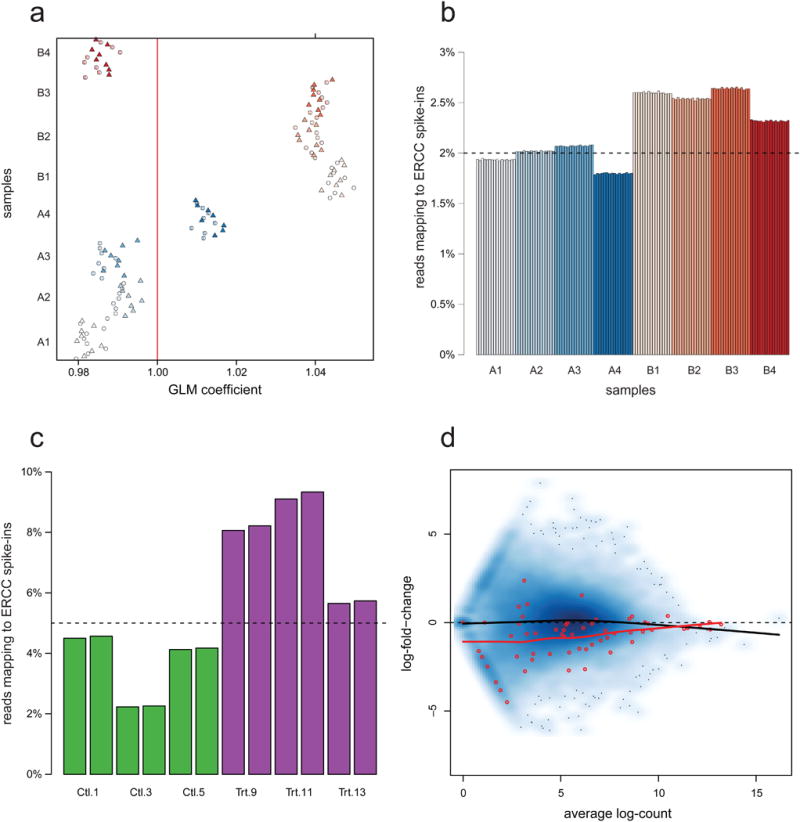
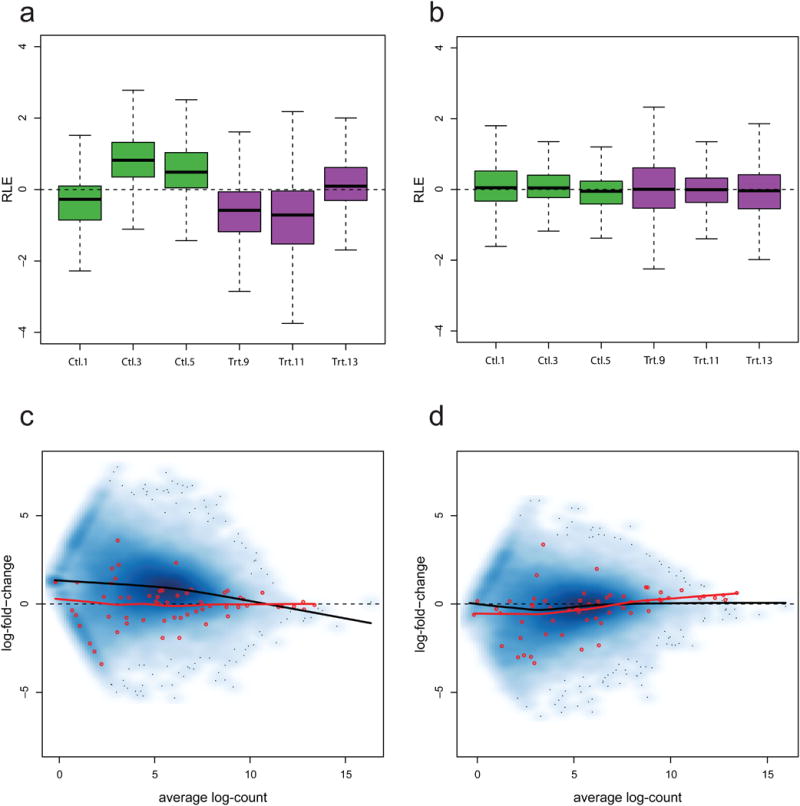
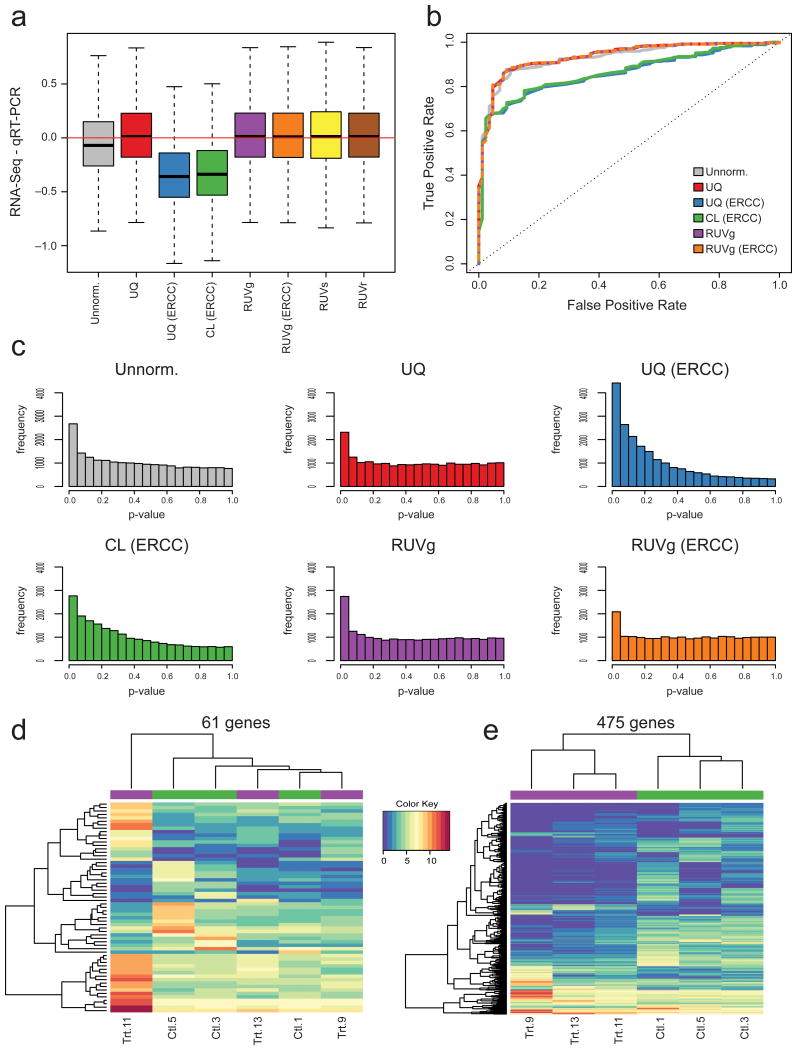
Normalization of RNA-sequencing (RNA-seq) data has proven essential to ensure accurate inference of expression levels. Here, we show that usual normalization approaches mostly account for sequencing depth and fail to correct for library preparation and other more complex unwanted technical effects. We evaluate the performance of the External RNA Control Consortium (ERCC) spike-in controls and investigate the possibility of using them directly for normalization. We show that the spike-ins are not reliable enough to be used in standard global-scaling or regression-based normalization procedures. We propose a normalization strategy, called remove unwanted variation (RUV), that adjusts for nuisance technical effects by performing factor analysis on suitable sets of control genes (e.g., ERCC spike-ins) or samples (e.g., replicate libraries). Our approach leads to more accurate estimates of expression fold-changes and tests of differential expression compared to state-of-the-art normalization methods. In particular, RUV promises to be valuable for large collaborative projects involving multiple laboratories, technicians, and/or sequencing platforms.
Figures
References
-
- Dillies MA, et al. A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Briefings in Bioinformatics. 2013;14:671–683. - PubMed
Publication types
MeSH terms
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
