

Regulation of alternative VEGF-A mRNA splicing is a therapeutic target for analgesia
- PMID: 25151644
- PMCID: PMC4194316
- DOI: 10.1016/j.nbd.2014.08.012
Regulation of alternative VEGF-A mRNA splicing is a therapeutic target for analgesia
Abstract
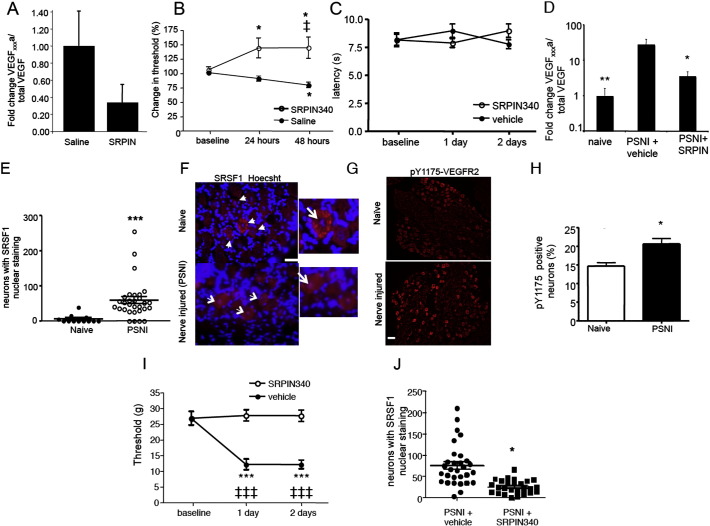
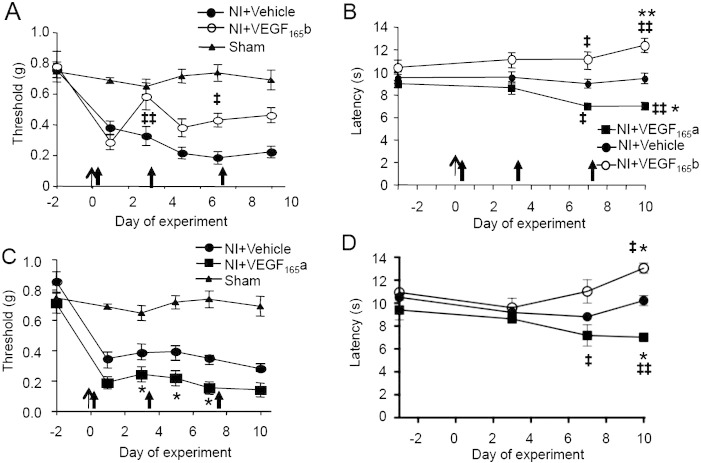
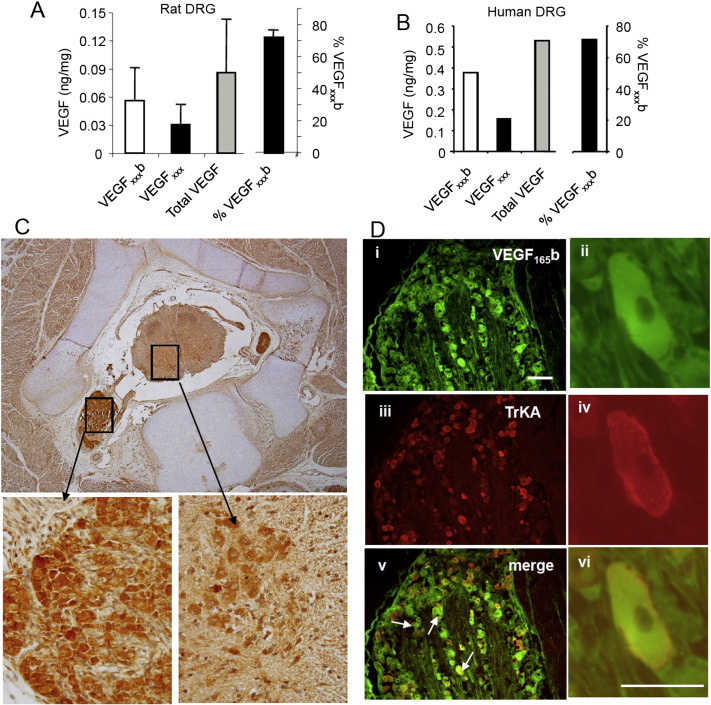
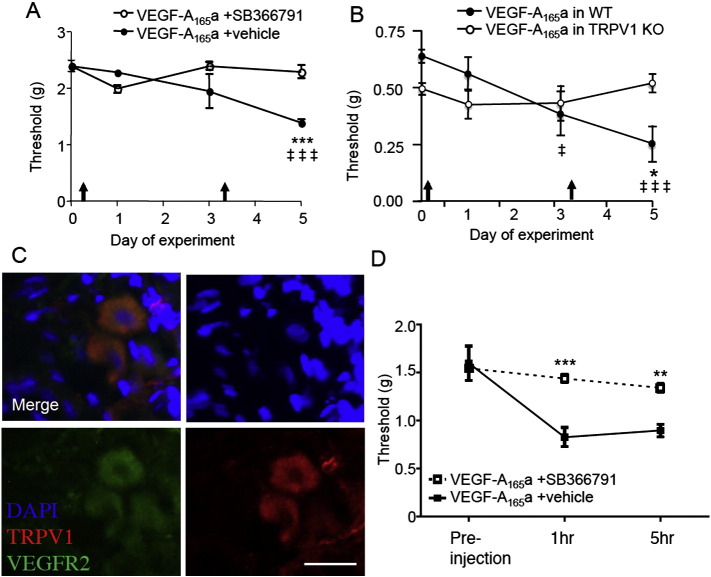
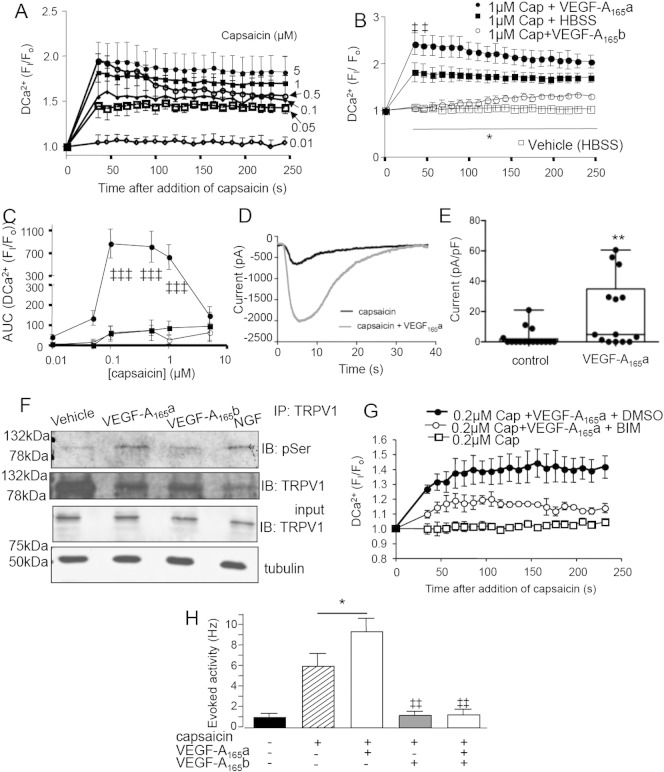
Vascular endothelial growth factor-A (VEGF-A) is best known as a key regulator of the formation of new blood vessels. Neutralization of VEGF-A with anti-VEGF therapy e.g. bevacizumab, can be painful, and this is hypothesized to result from a loss of VEGF-A-mediated neuroprotection. The multiple vegf-a gene products consist of two alternatively spliced families, typified by VEGF-A165a and VEGF-A165b (both contain 165 amino acids), both of which are neuroprotective. Under pathological conditions, such as in inflammation and cancer, the pro-angiogenic VEGF-A165a is upregulated and predominates over the VEGF-A165b isoform. We show here that in rats and mice VEGF-A165a and VEGF-A165b have opposing effects on pain, and that blocking the proximal splicing event - leading to the preferential expression of VEGF-A165b over VEGF165a - prevents pain in vivo. VEGF-A165a sensitizes peripheral nociceptive neurons through actions on VEGFR2 and a TRPV1-dependent mechanism, thus enhancing nociceptive signaling. VEGF-A165b blocks the effect of VEGF-A165a. After nerve injury, the endogenous balance of VEGF-A isoforms switches to greater expression of VEGF-Axxxa compared to VEGF-Axxxb, through an SRPK1-dependent pre-mRNA splicing mechanism. Pharmacological inhibition of SRPK1 after traumatic nerve injury selectively reduced VEGF-Axxxa expression and reversed associated neuropathic pain. Exogenous VEGF-A165b also ameliorated neuropathic pain. We conclude that the relative levels of alternatively spliced VEGF-A isoforms are critical for pain modulation under both normal conditions and in sensory neuropathy. Altering VEGF-Axxxa/VEGF-Axxxb balance by targeting alternative RNA splicing may be a new analgesic strategy.
Keywords: Alternative mRNA splicing; Neuropathy; Nociceptors; Vascular endothelial growth factor A.
Copyright © 2014. Published by Elsevier Inc.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
