

SubcloneSeeker: a computational framework for reconstructing tumor clone structure for cancer variant interpretation and prioritization
- PMID: 25160522
- PMCID: PMC4180956
- DOI: 10.1186/s13059-014-0443-x
SubcloneSeeker: a computational framework for reconstructing tumor clone structure for cancer variant interpretation and prioritization
Abstract
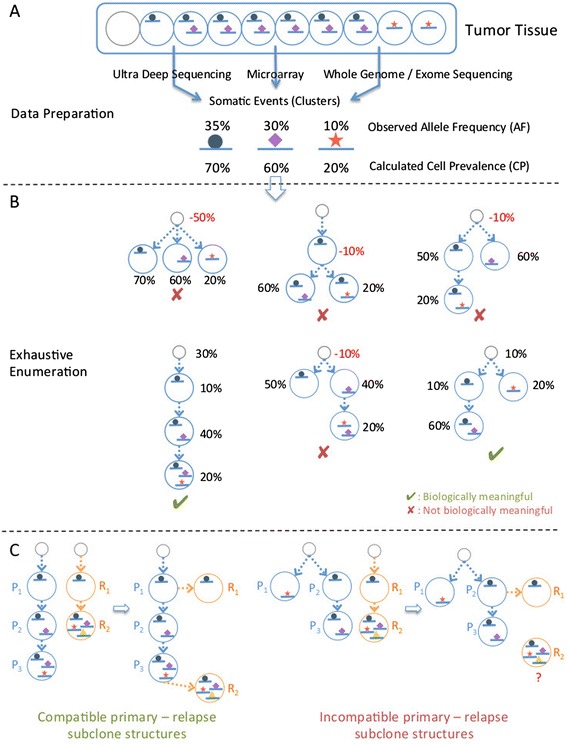
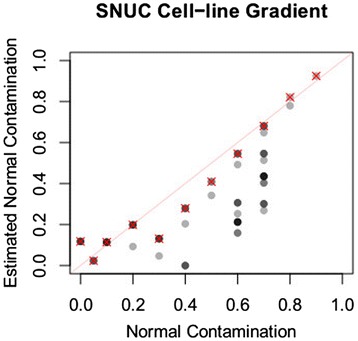
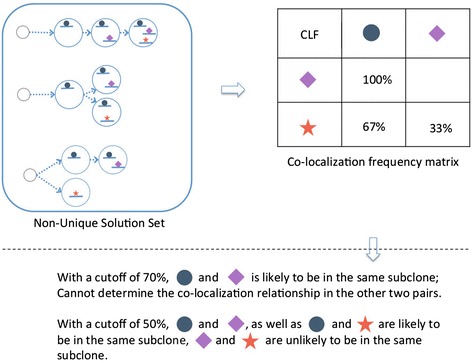
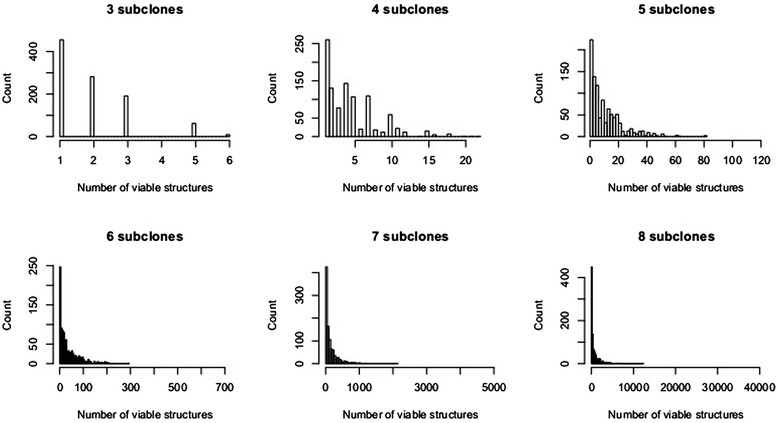
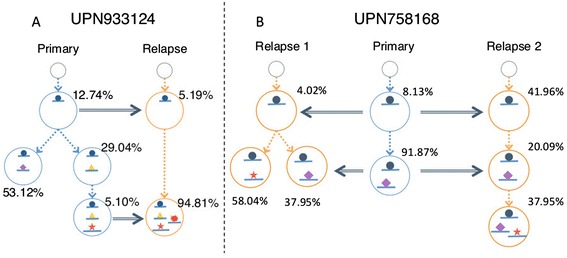
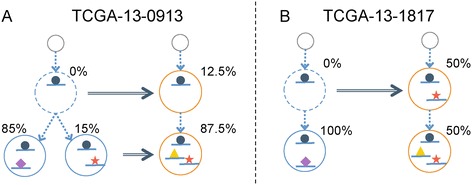
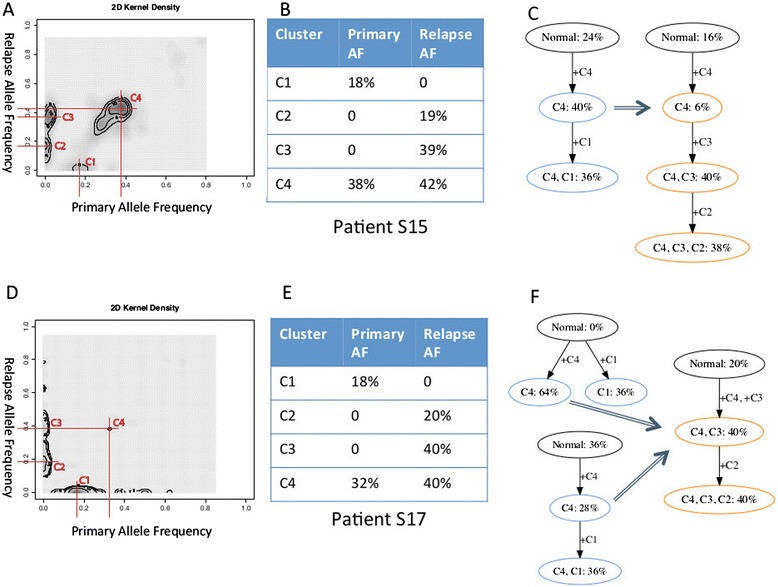
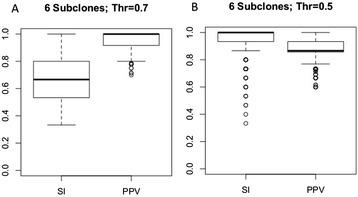
Many tumors are composed of genetically divergent cell subpopulations. We report SubcloneSeeker, a package capable of exhaustive identification of subclone structures and evolutionary histories with bulk somatic variant allele frequency measurements from tumor biopsies. We present a statistical framework to elucidate whether specific sets of mutations are present within the same subclones, and the order in which they occur. We demonstrate how subclone reconstruction provides crucial information about tumorigenesis and relapse mechanisms; guides functional study by variant prioritization, and has the potential as a rational basis for informed therapeutic strategies for the patient. SubcloneSeeker is available at: https://github.com/yiq/SubcloneSeeker.
Figures
References
-
- Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, Varela I, Lin ML, Ordonez GR, Bignell GR, Ye K, Alipaz J, Bauer MJ, Beare D, Butler A, Carter RJ, Chen L, Cox AJ, Edkins S, Kokko-Gonzales PI, Gormley NA, Grocock RJ, Haudenschild CD, Hims MM, James T, Jia M, Kingsbury Z, Leroy C, Marshall J, Menzies A, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–196. doi: 10.1038/nature08658. - DOI - PMC - PubMed
-
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, Kiezun A, Hammerman PS, McKenna A, Drier Y, Zou L, Ramos AH, Pugh TJ, Stransky N, Helman E, Kim J, Sougnez C, Ambrogio L, Nickerson E, Shefler E, Cortes ML, Auclair D, Saksena G, Voet D, Noble M, DiCara D, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. - DOI - PMC - PubMed
-
- Gonzalez-Perez A, Mustonen V, Reva B, Ritchie GR, Creixell P, Karchin R, Vazquez M, Fink JL, Kassahn KS, Pearson JV, Bader GD, Boutros PC, Muthuswamy L, Ouellette BF, Reimand J, Linding R, Shibata T, Valencia A, Butler A, Dronov S, Flicek P, Shannon NB, Carter H, Ding L, Sander C, Stuart JM, Stein LD, Lopez-Bigas N, International Cancer Genome Consortium Mutation P, Consequences Subgroup of the Bioinformatics Analyses Working G Computational approaches to identify functional genetic variants in cancer genomes. Nat Methods. 2013;10:723–729. doi: 10.1038/nmeth.2562. - DOI - PMC - PubMed
-
- Keats JJ, Chesi M, Egan JB, Garbitt VM, Palmer SE, Braggio E, Van Wier S, Blackburn PR, Baker AS, Dispenzieri A, Kumar S, Rajkumar SV, Carpten JD, Barrett M, Fonseca R, Stewart AK, Bergsagel PL. Clonal competition with alternating dominance in multiple myeloma. Blood. 2012;120:1067–1076. doi: 10.1182/blood-2012-01-405985. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
