

Human-specific bacterial pore-forming toxins induce programmed necrosis in erythrocytes
- PMID: 25161188
- PMCID: PMC4173772
- DOI: 10.1128/mBio.01251-14
Human-specific bacterial pore-forming toxins induce programmed necrosis in erythrocytes
Abstract
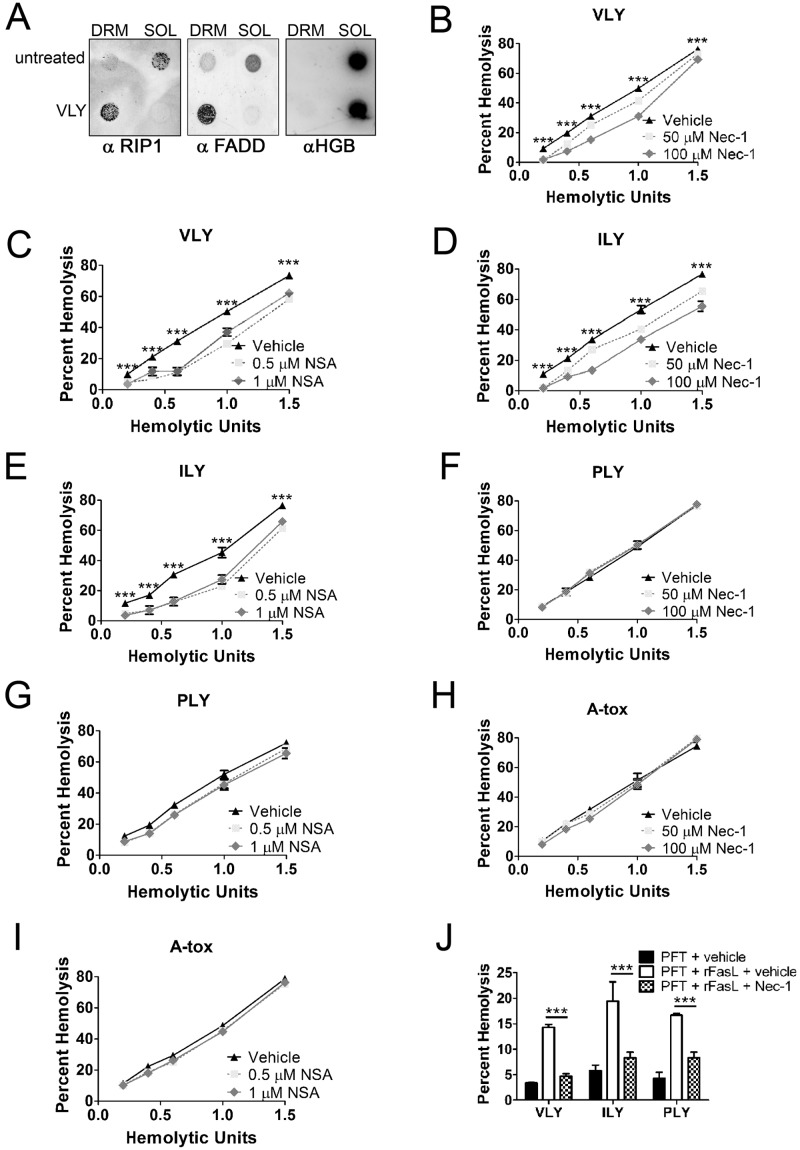
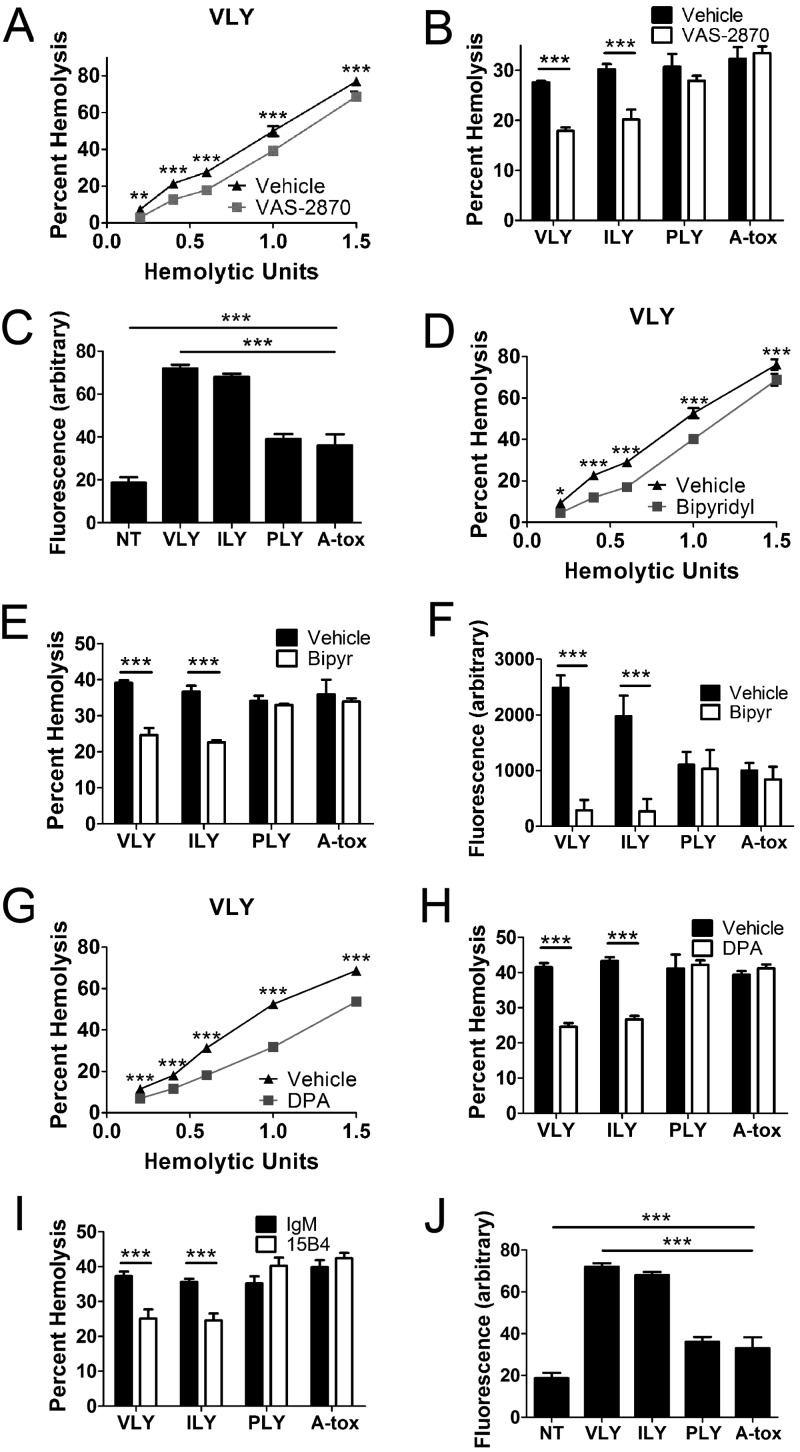
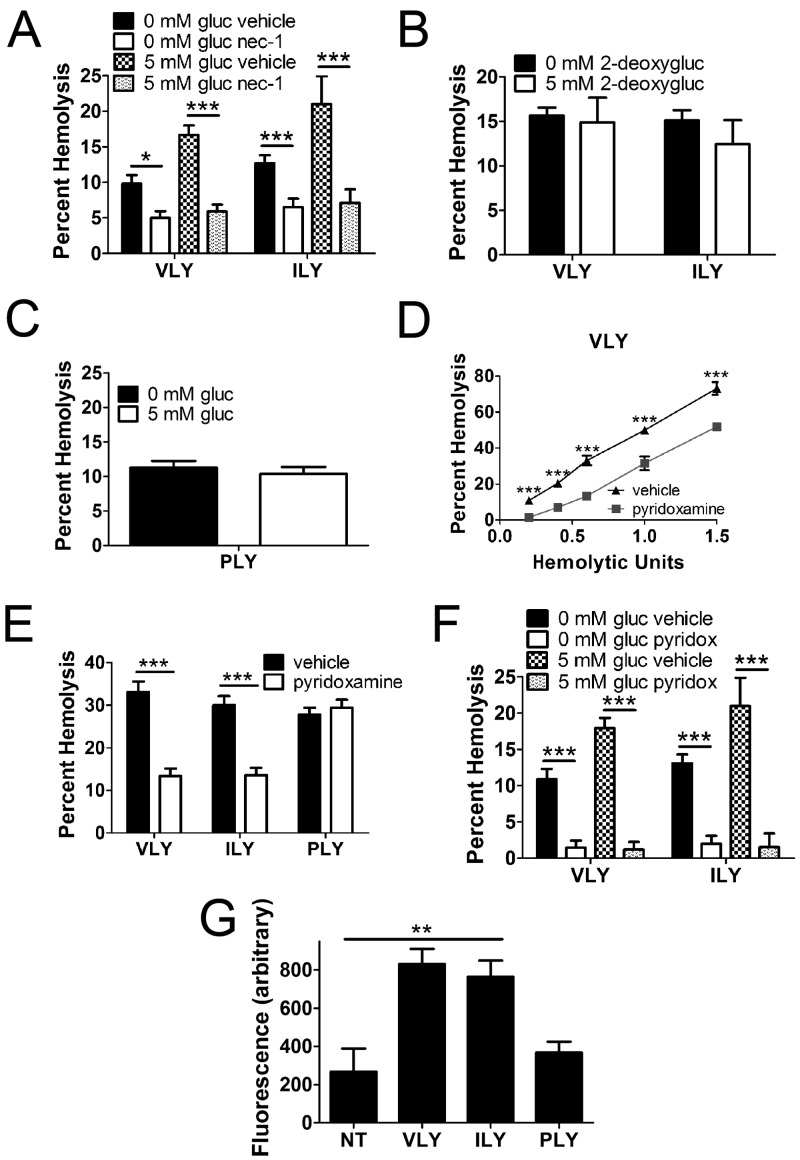
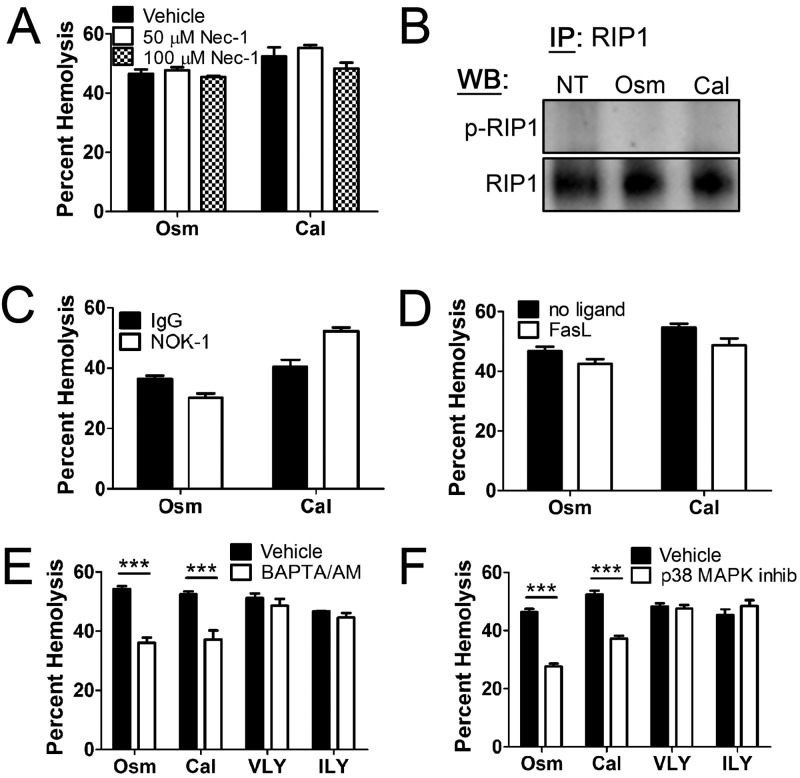
A subgroup of the cholesterol-dependent cytolysin (CDC) family of pore-forming toxins (PFTs) has an unusually narrow host range due to a requirement for binding to human CD59 (hCD59), a glycosylphosphatidylinositol (GPI)-linked complement regulatory molecule. hCD59-specific CDCs are produced by several organisms that inhabit human mucosal surfaces and can act as pathogens, including Gardnerella vaginalis and Streptococcus intermedius. The consequences and potential selective advantages of such PFT host limitation have remained unknown. Here, we demonstrate that, in addition to species restriction, PFT ligation of hCD59 triggers a previously unrecognized pathway for programmed necrosis in primary erythrocytes (red blood cells [RBCs]) from humans and transgenic mice expressing hCD59. Because they lack nuclei and mitochondria, RBCs have typically been thought to possess limited capacity to undergo programmed cell death. RBC programmed necrosis shares key molecular factors with nucleated cell necroptosis, including dependence on Fas/FasL signaling and RIP1 phosphorylation, necrosome assembly, and restriction by caspase-8. Death due to programmed necrosis in RBCs is executed by acid sphingomyelinase-dependent ceramide formation, NADPH oxidase- and iron-dependent reactive oxygen species formation, and glycolytic formation of advanced glycation end products. Bacterial PFTs that are hCD59 independent do not induce RBC programmed necrosis. RBC programmed necrosis is biochemically distinct from eryptosis, the only other known programmed cell death pathway in mature RBCs. Importantly, RBC programmed necrosis enhances the growth of PFT-producing pathogens during exposure to primary RBCs, consistent with a role for such signaling in microbial growth and pathogenesis.
Importance: In this work, we provide the first description of a new form of programmed cell death in erythrocytes (RBCs) that occurs as a consequence of cellular attack by human-specific bacterial toxins. By defining a new RBC death pathway that shares important components with necroptosis, a programmed necrosis module that occurs in nucleated cells, these findings expand our understanding of RBC biology and RBC-pathogen interactions. In addition, our work provides a link between cholesterol-dependent cytolysin (CDC) host restriction and promotion of bacterial growth in the presence of RBCs, which may provide a selective advantage to human-associated bacterial strains that elaborate such toxins and a potential explanation for the narrowing of host range observed in this toxin family.
Copyright © 2014 LaRocca et al.
Figures





References
-
- Ucar K. 2002. Clinical presentation and management of hemolytic anemias. Oncology (Williston Park) 16(9 Suppl 10):163–170 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
