

Viral pathogen-associated molecular patterns regulate blood-brain barrier integrity via competing innate cytokine signals
- PMID: 25161189
- PMCID: PMC4173776
- DOI: 10.1128/mBio.01476-14
Viral pathogen-associated molecular patterns regulate blood-brain barrier integrity via competing innate cytokine signals
Abstract
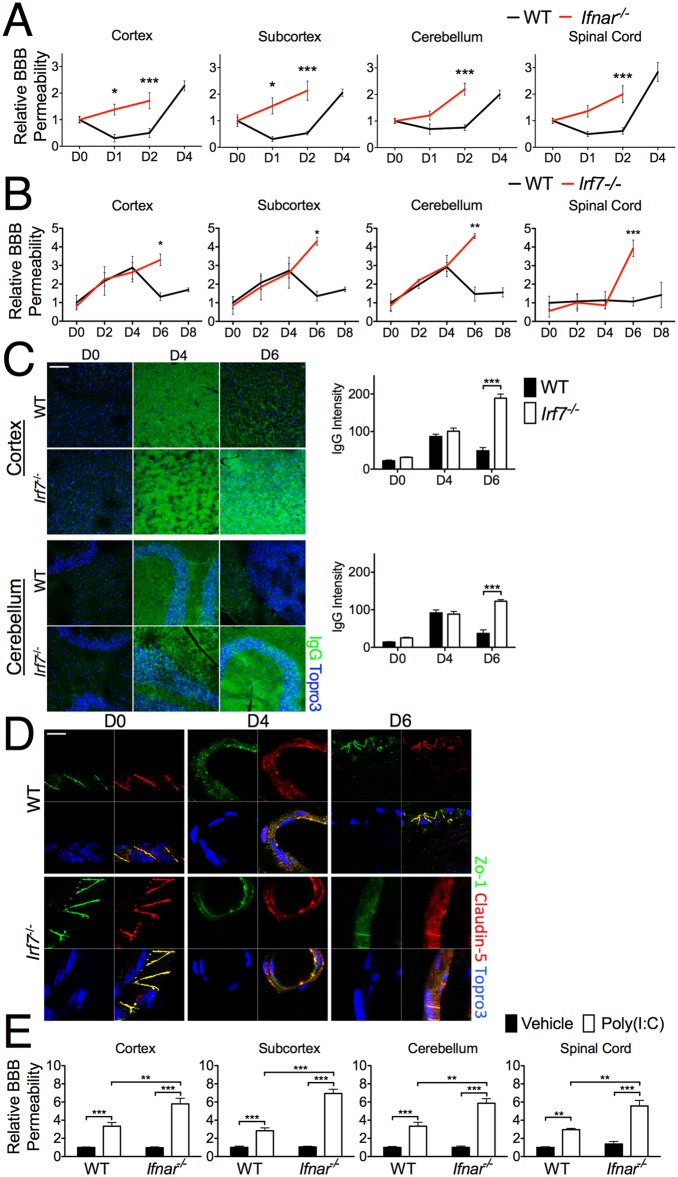
Pattern recognition receptor (PRR) detection of pathogen-associated molecular patterns (PAMPs), such as viral RNA, drives innate immune responses against West Nile virus (WNV), an emerging neurotropic pathogen. Here we demonstrate that WNV PAMPs orchestrate endothelial responses to WNV via competing innate immune cytokine signals at the blood-brain barrier (BBB), a multicellular interface with highly specialized brain endothelial cells that normally prevents pathogen entry. While Th1 cytokines increase the permeability of endothelial barriers, type I interferon (IFN) promoted and stabilized BBB function. Induction of innate cytokines by pattern recognition pathways directly regulated BBB permeability and tight junction formation via balanced activation of the small GTPases Rac1 and RhoA, which in turn regulated the transendothelial trafficking of WNV. In vivo, mice with attenuated type I IFN signaling or IFN induction (Ifnar(-/-) Irf7(-/-)) exhibited enhanced BBB permeability and tight junction dysregulation after WNV infection. Together, these data provide new insight into host-pathogen interactions at the BBB during neurotropic viral infection.
Importance: West Nile virus (WNV) is an emerging pathogen capable of infecting the central nervous system (CNS), causing fatal encephalitis. However, the mechanisms that control the ability of WNV to cross the blood-brain barrier (BBB) and access the CNS are unclear. In this study, we show that detection of WNV by host tissues induces innate immune cytokine expression at the BBB, regulating BBB structure and function and impacting transendothelial trafficking of WNV. This regulatory effect is shown to happen rapidly following exposure to virus, to occur independently of viral replication within BBB cells, and to require the signaling of cytoskeletal regulatory Rho GTPases. These results provide new understanding of host-pathogen interactions at the BBB during viral encephalitis.
Copyright © 2014 Daniels et al.
Figures




References
-
- Verma S, Lo Y, Chapagain M, Lum S, Kumar M, Gurjav U, Luo H, Nakatsuka A, Nerurkar VR. 2009. West Nile virus infection modulates human brain microvascular endothelial cells tight junction proteins and cell adhesion molecules: transmigration across the in vitro blood-brain barrier. Virology 385:425–433. 10.1016/j.virol.2008.11.047 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
