

Comparative analysis of regulatory information and circuits across distant species
- PMID: 25164757
- PMCID: PMC4336544
- DOI: 10.1038/nature13668
Comparative analysis of regulatory information and circuits across distant species
Abstract
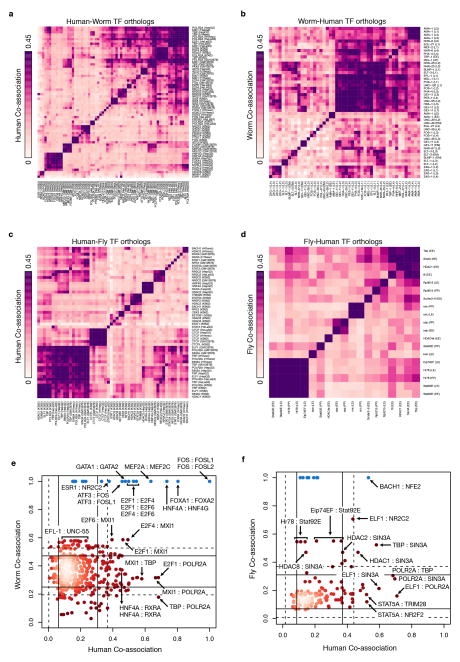
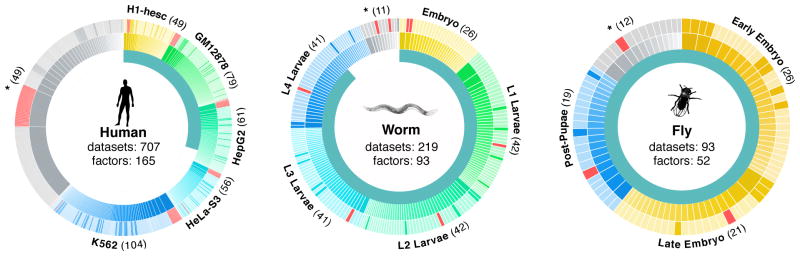
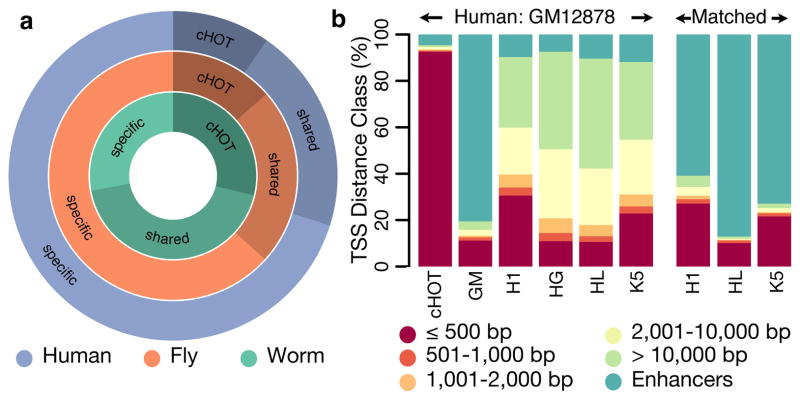
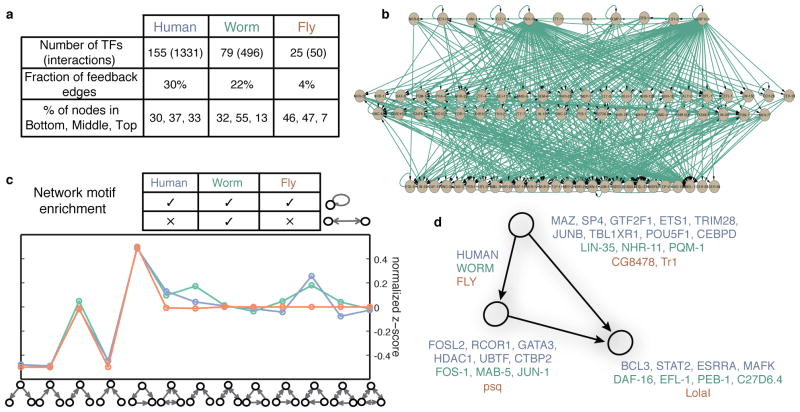
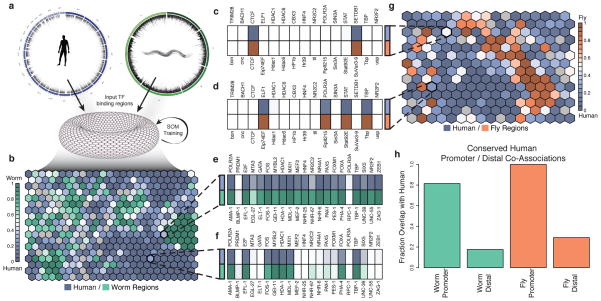
Despite the large evolutionary distances between metazoan species, they can show remarkable commonalities in their biology, and this has helped to establish fly and worm as model organisms for human biology. Although studies of individual elements and factors have explored similarities in gene regulation, a large-scale comparative analysis of basic principles of transcriptional regulatory features is lacking. Here we map the genome-wide binding locations of 165 human, 93 worm and 52 fly transcription regulatory factors, generating a total of 1,019 data sets from diverse cell types, developmental stages, or conditions in the three species, of which 498 (48.9%) are presented here for the first time. We find that structural properties of regulatory networks are remarkably conserved and that orthologous regulatory factor families recognize similar binding motifs in vivo and show some similar co-associations. Our results suggest that gene-regulatory properties previously observed for individual factors are general principles of metazoan regulation that are remarkably well-preserved despite extensive functional divergence of individual network connections. The comparative maps of regulatory circuitry provided here will drive an improved understanding of the regulatory underpinnings of model organism biology and how these relate to human biology, development and disease.
Conflict of interest statement
MPS is a cofounder and scientific advisory board (SAB) member of Personalis. MPS is on the SAB of Genapsys.
Figures
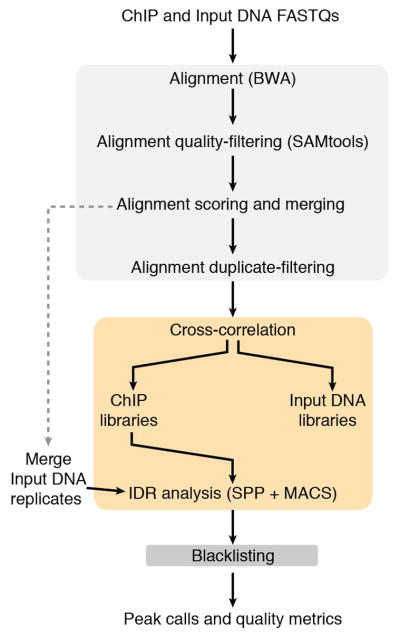
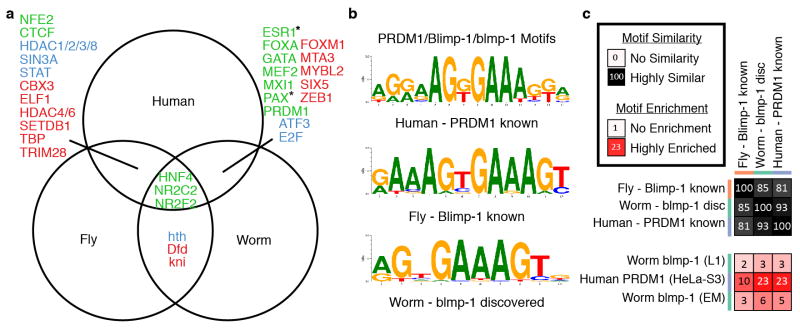
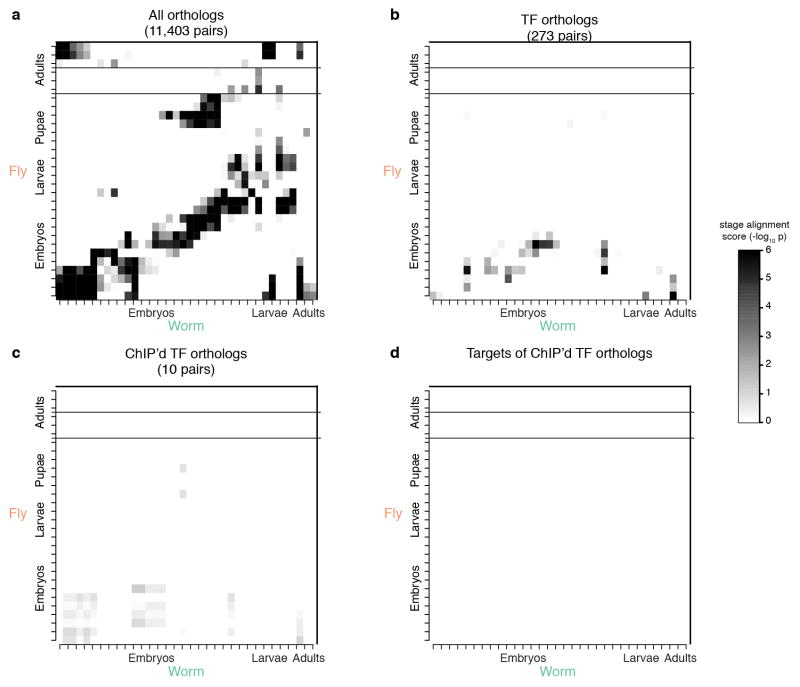
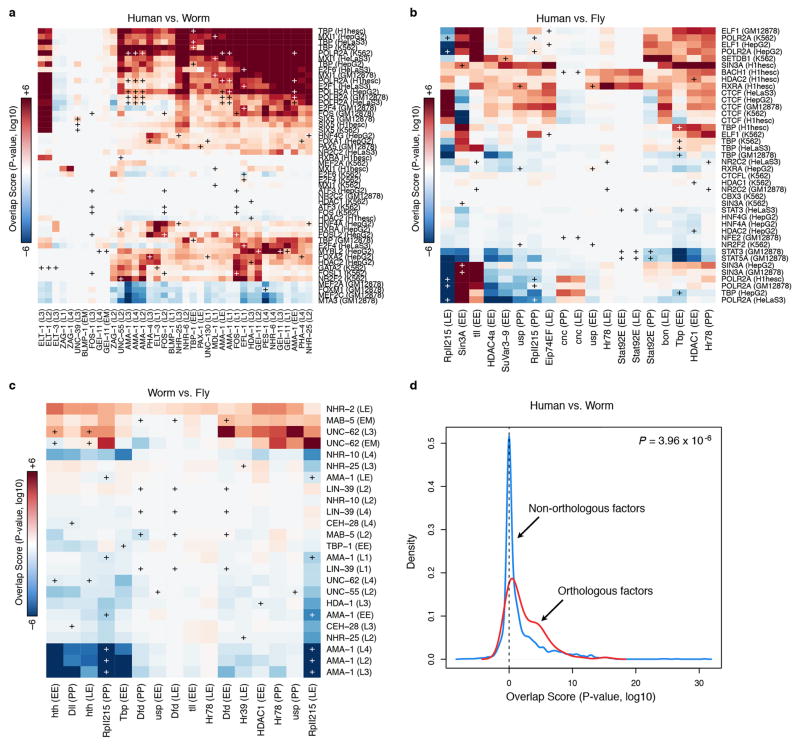
Comment in
-
Genomics: Hiding in plain sight.Nature. 2014 Aug 28;512(7515):374-5. doi: 10.1038/512374a. Nature. 2014. PMID: 25164742 No abstract available.
References
-
- Gerstein M, et al. An Integrative Comparison of Metazoan Transcriptomes
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
