

Stepwise priming by acidic pH and a high K+ concentration is required for efficient uncoating of influenza A virus cores after penetration
- PMID: 25165113
- PMCID: PMC4249060
- DOI: 10.1128/JVI.01430-14
Stepwise priming by acidic pH and a high K+ concentration is required for efficient uncoating of influenza A virus cores after penetration
Abstract
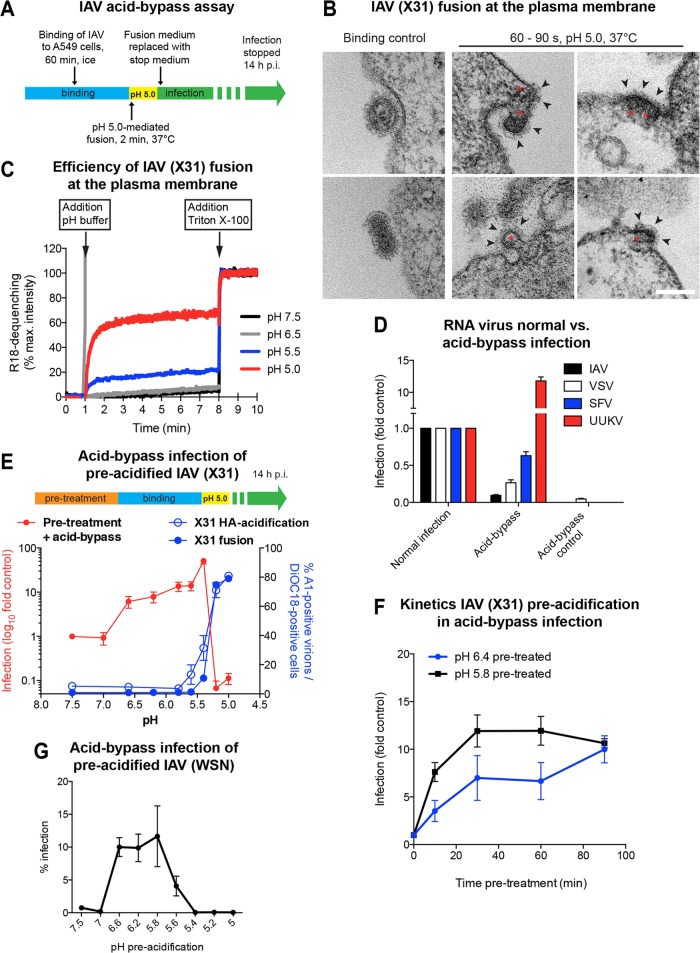
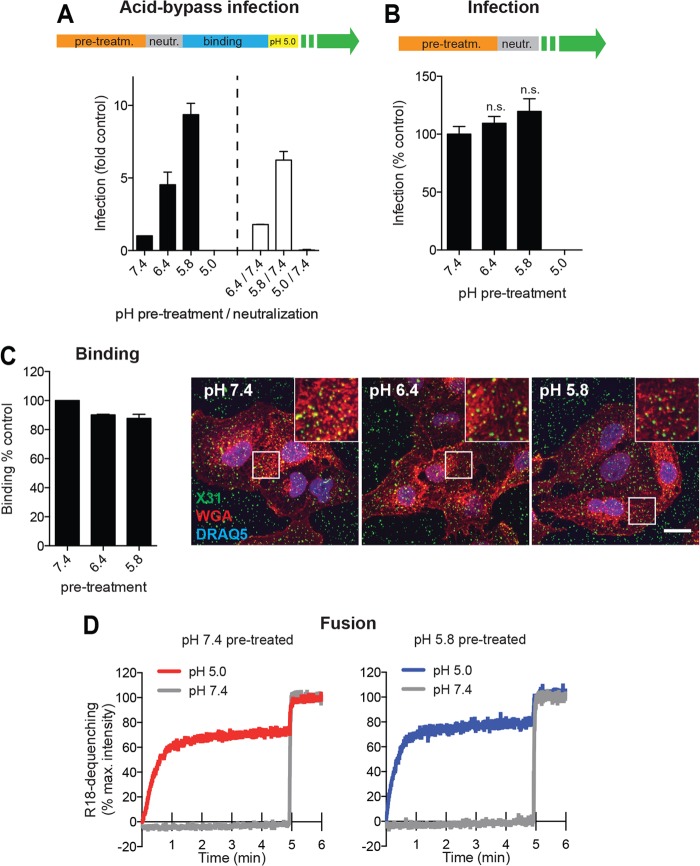
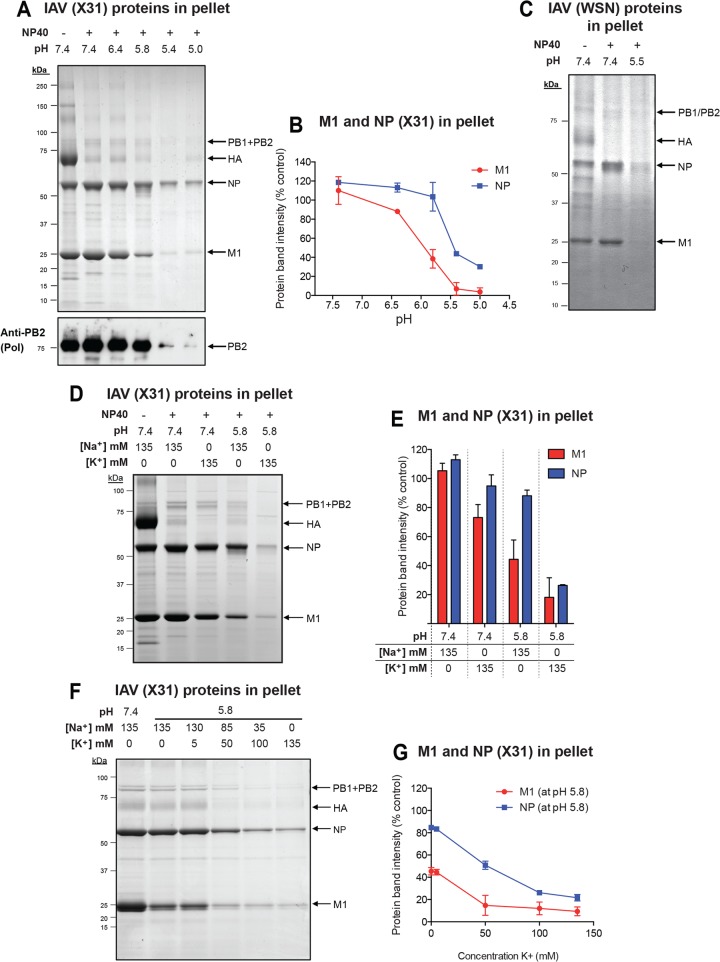
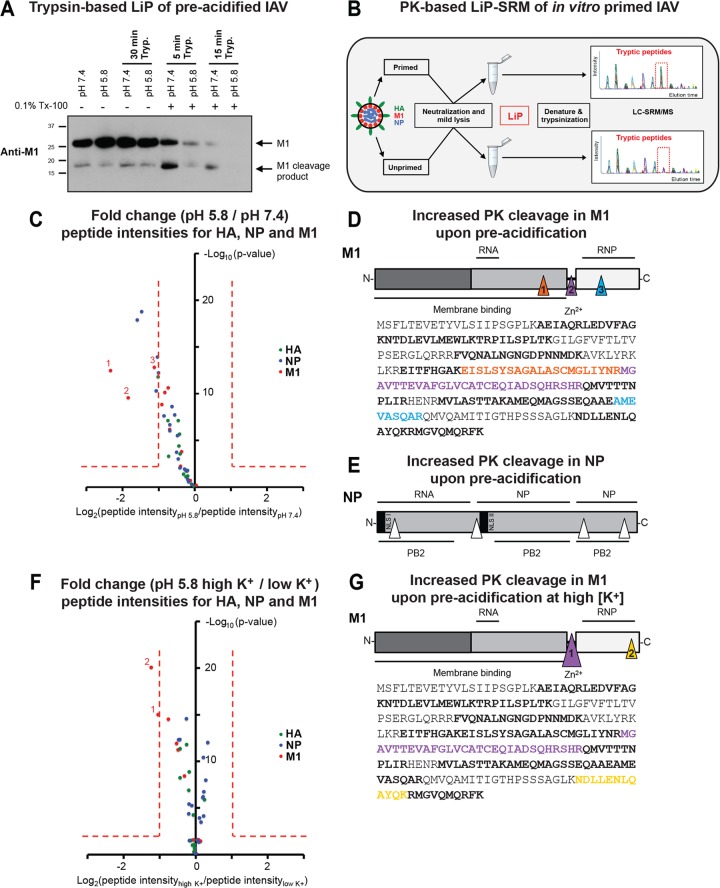
Influenza A virus (IAV) uses the low pH in late endocytic vacuoles as a cue for penetration by membrane fusion. Here, we analyzed the prefusion reactions that prepare the core for uncoating after it has been delivered to the cytosol. We found that this priming process occurs in two steps that are mediated by the envelope-embedded M2 ion channel. The first weakens the interactions between the matrix protein, M1, and the viral ribonucleoprotein bundle. It involves a conformational change in a linker sequence and the C-terminal domain of M1 after exposure to a pH below 6.5. The second step is triggered by a pH of <6.0 and by the influx of K(+) ions. It causes additional changes in M1 as well as a loss of stability in the viral ribonucleoprotein bundle. Our results indicate that both the switch from Na(+) to K(+) in maturing endosomes and the decreasing pH are needed to prime IAV cores for efficient uncoating and infection of the host cell.
Importance: The entry of IAV involves several steps, including endocytosis and fusion at late endosomes. Entry also includes disassembly of the viral core, which is composed of the viral ribonucleoproteins and the RNA genome. We have found that the uncoating process of IAV is initiated long before the core is delivered into the cytosol. M2, an ion channel in the viral membrane, is activated when the virus passes through early endosomes. Here, we show that protons entering the virus through M2 cause a conformational change in the matrix protein, M1. This weakens interactions between M1 and the viral ribonucleoproteins. A second change was found to occur when the virus enters late endosomes. The preacidified core is then exposed to a high concentration of K(+), which affects the interactions between the ribonucleoproteins. Thus, when cores are finally delivered to the cytosol, they are already partially destabilized and, therefore, uncoating competent and infectious.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
Figures


References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
