

Measuring the activity of protein variants on a large scale using deep mutational scanning
- PMID: 25167058
- PMCID: PMC4412028
- DOI: 10.1038/nprot.2014.153
Measuring the activity of protein variants on a large scale using deep mutational scanning
Abstract
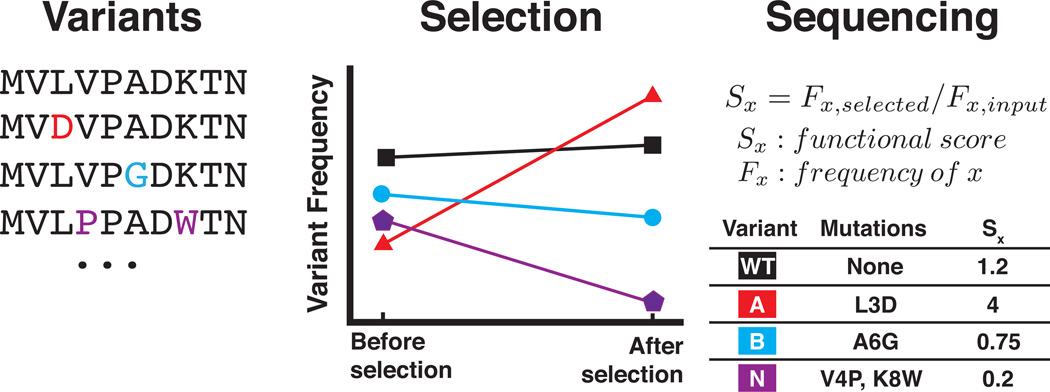
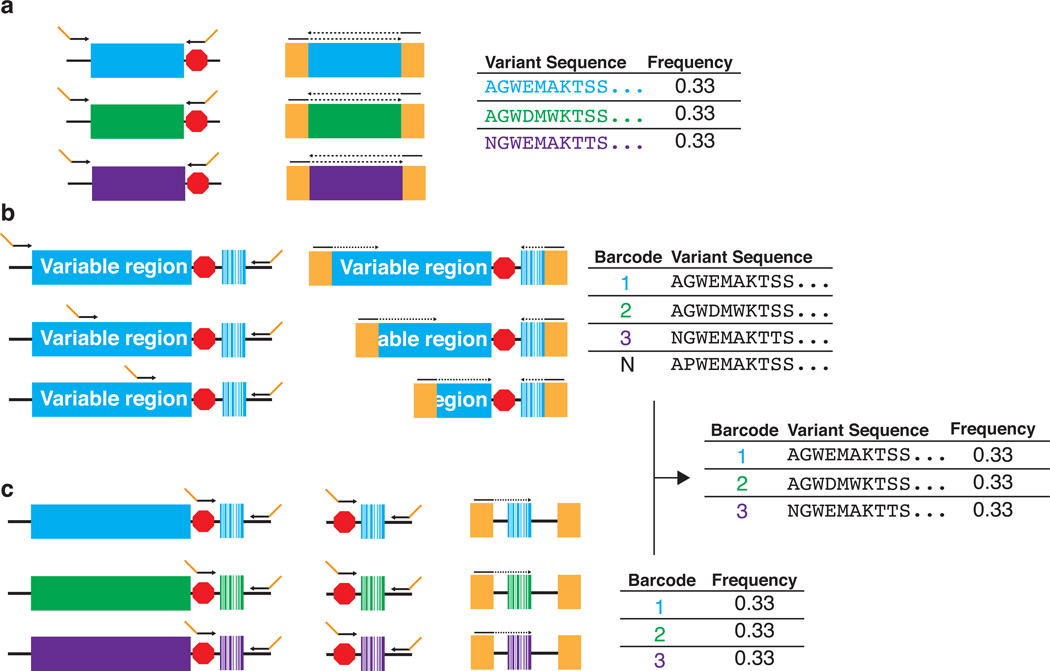
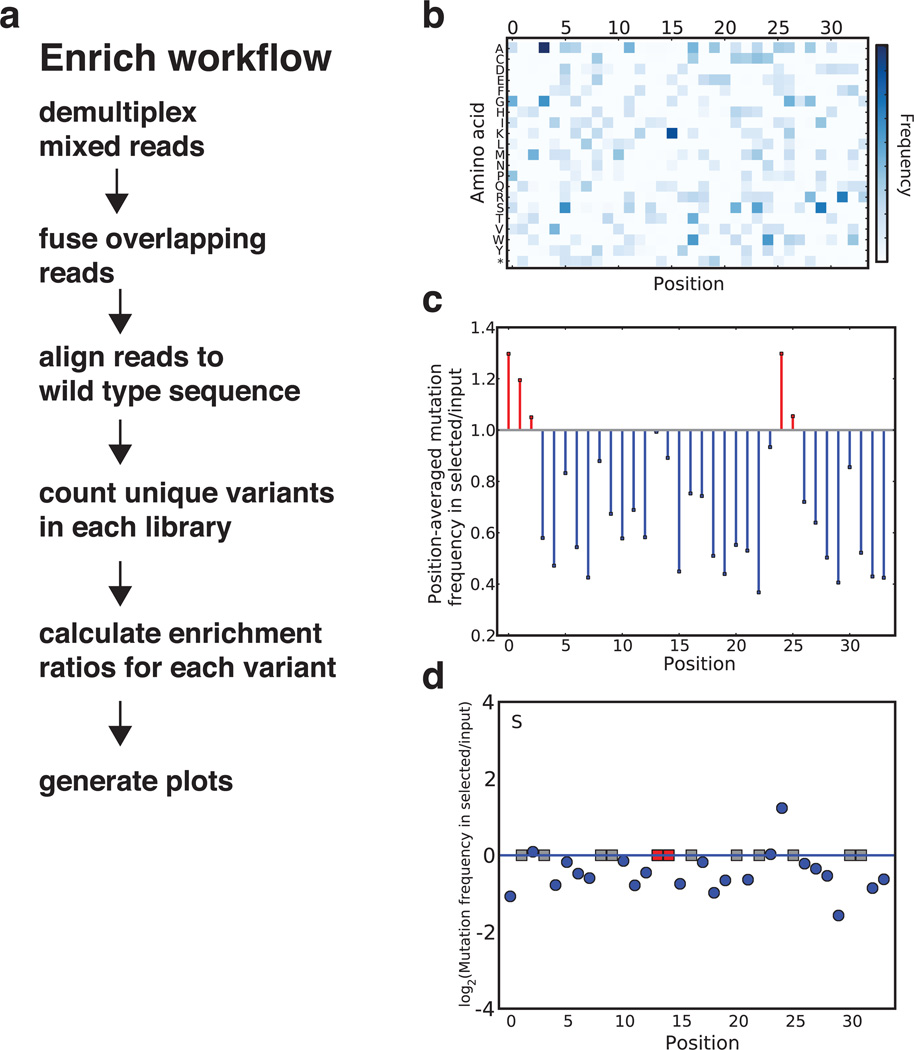
Deep mutational scanning marries selection for protein function to high-throughput DNA sequencing in order to quantify the activity of variants of a protein on a massive scale. First, an appropriate selection system for the protein function of interest is identified and validated. Second, a library of variants is created, introduced into the selection system and subjected to selection. Third, library DNA is recovered throughout the selection and deep-sequenced. Finally, a functional score for each variant is calculated on the basis of the change in the frequency of the variant during the selection. This protocol describes the steps that must be carried out to generate a large-scale mutagenesis data set consisting of functional scores for up to hundreds of thousands of variants of a protein of interest. Establishing an assay, generating a library of variants and carrying out a selection and its accompanying sequencing takes on the order of 4-6 weeks; the initial data analysis can be completed in 1 week.
Figures

References
-
- Cunningham BC, Wells JA. High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science. 1989;244:1081–1085. - PubMed
-
- Pál G, Kouadio JLK, Artis DR, Kossiakoff AA, Sidhu SS. Comprehensive and quantitative mapping of energy landscapes for protein-protein interactions by rapid combinatorial scanning. J. Biol. Chem. 2006;281:22378–22385. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
