

Mutations in IFT172 cause isolated retinal degeneration and Bardet-Biedl syndrome
- PMID: 25168386
- PMCID: PMC4326328
- DOI: 10.1093/hmg/ddu441
Mutations in IFT172 cause isolated retinal degeneration and Bardet-Biedl syndrome
Abstract
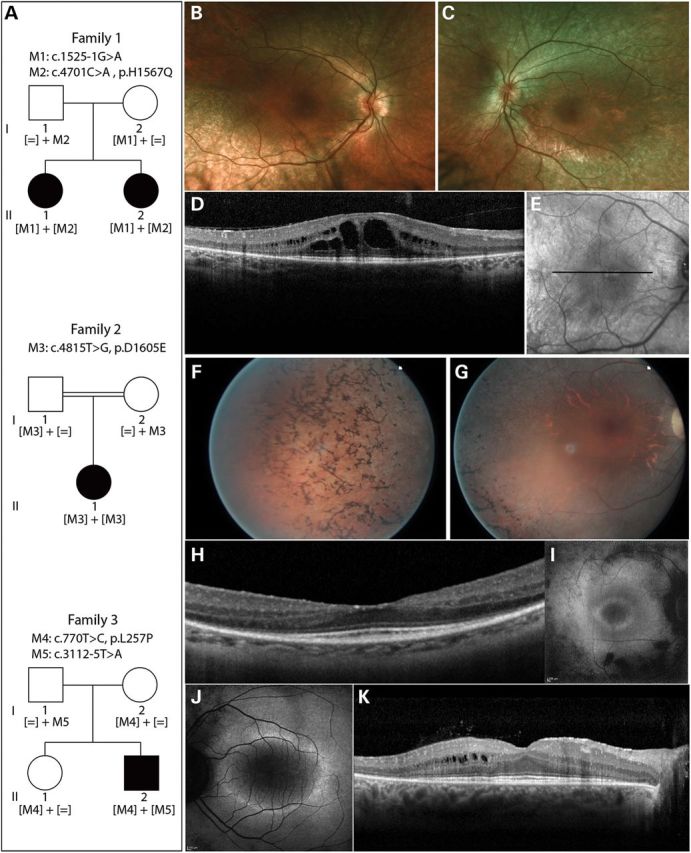
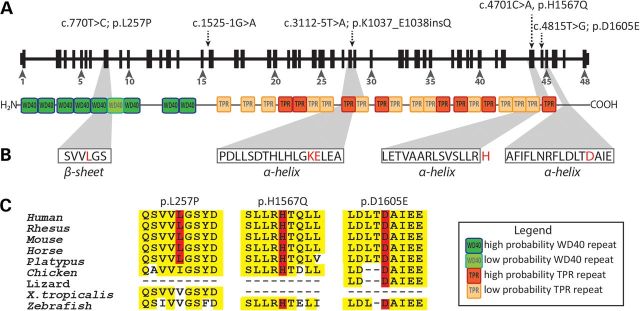
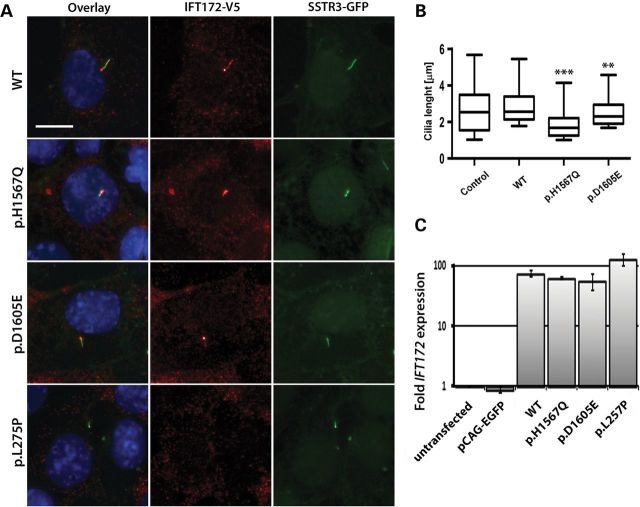
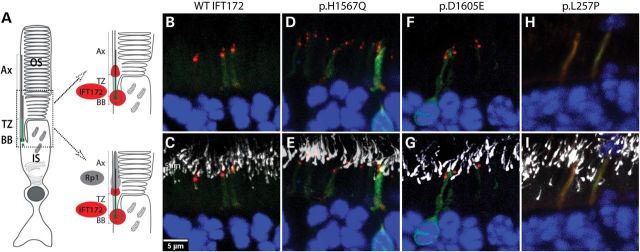
Primary cilia are sensory organelles present on most mammalian cells. The assembly and maintenance of primary cilia are facilitated by intraflagellar transport (IFT), a bidirectional protein trafficking along the cilium. Mutations in genes coding for IFT components have been associated with a group of diseases called ciliopathies. These genetic disorders can affect a variety of organs including the retina. Using whole exome sequencing in three families, we identified mutations in Intraflagellar Transport 172 Homolog [IFT172 (Chlamydomonas)] that underlie an isolated retinal degeneration and Bardet-Biedl syndrome. Extensive functional analyses of the identified mutations in cell culture, rat retina and in zebrafish demonstrated their hypomorphic or null nature. It has recently been reported that mutations in IFT172 cause a severe ciliopathy syndrome involving skeletal, renal, hepatic and retinal abnormalities (Jeune and Mainzer-Saldino syndromes). Here, we report for the first time that mutations in this gene can also lead to an isolated form of retinal degeneration. The functional data for the mutations can partially explain milder phenotypes; however, the involvement of modifying alleles in the IFT172-associated phenotypes cannot be excluded. These findings expand the spectrum of disease associated with mutations in IFT172 and suggest that mutations in genes originally reported to be associated with syndromic ciliopathies should also be considered in subjects with non-syndromic retinal dystrophy.
© The Author 2014. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
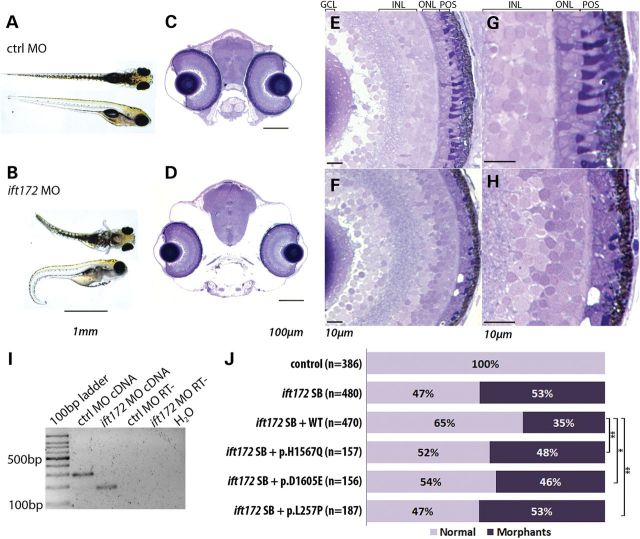
References
-
- Mockel A., Perdomo Y., Stutzmann F., Letsch J., Marion V., Dollfus H. Retinal dystrophy in Bardet–Biedl syndrome and related syndromic ciliopathies. Prog. Retin. Eye Res. 2011;30:258–274. - PubMed
-
- Pierce E.A., Quinn T., Meehan T., McGee T.L., Berson E.L., Dryja T.P. Mutations in a gene encoding a new oxygen-regulated photoreceptor protein cause dominant retinitis pigmentosa. Nat. Genet. 1999;22:248–254. - PubMed
-
- Guillonneau X., Piriev N.I., Danciger M., Kozak C.A., Cideciyan A.V., Jacobson S.G., Farber D.B. A nonsense mutation in a novel gene is associated with retinitis pigmentosa in a family linked to the RP1 locus. Hum. Mol. Genet. 1999;8:1541–1546. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
