

Characterization of the core and accessory genomes of Pseudomonas aeruginosa using bioinformatic tools Spine and AGEnt
- PMID: 25168460
- PMCID: PMC4155085
- DOI: 10.1186/1471-2164-15-737
Characterization of the core and accessory genomes of Pseudomonas aeruginosa using bioinformatic tools Spine and AGEnt
Abstract
Background: Pseudomonas aeruginosa is an important opportunistic pathogen responsible for many infections in hospitalized and immunocompromised patients. Previous reports estimated that approximately 10% of its 6.6 Mbp genome varies from strain to strain and is therefore referred to as "accessory genome". Elements within the accessory genome of P. aeruginosa have been associated with differences in virulence and antibiotic resistance. As whole genome sequencing of bacterial strains becomes more widespread and cost-effective, methods to quickly and reliably identify accessory genomic elements in newly sequenced P. aeruginosa genomes will be needed.
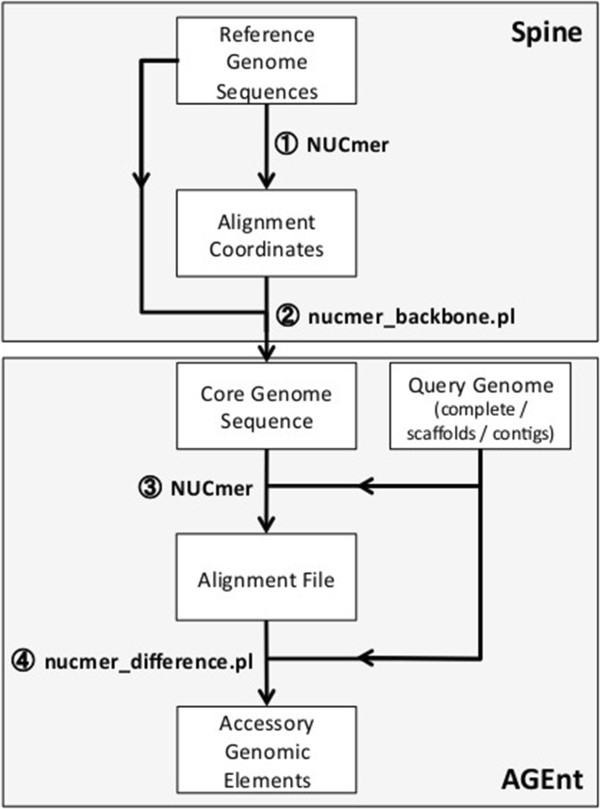
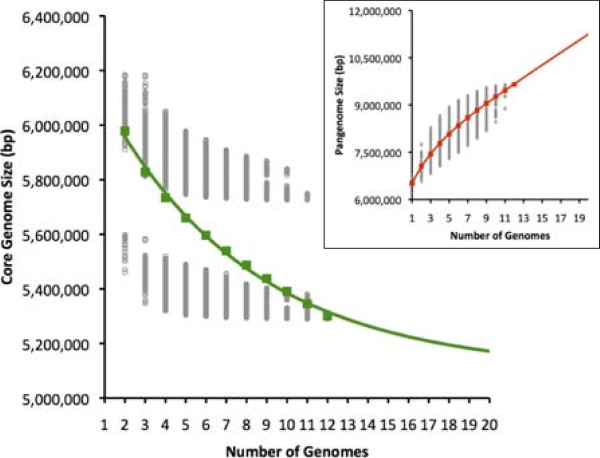
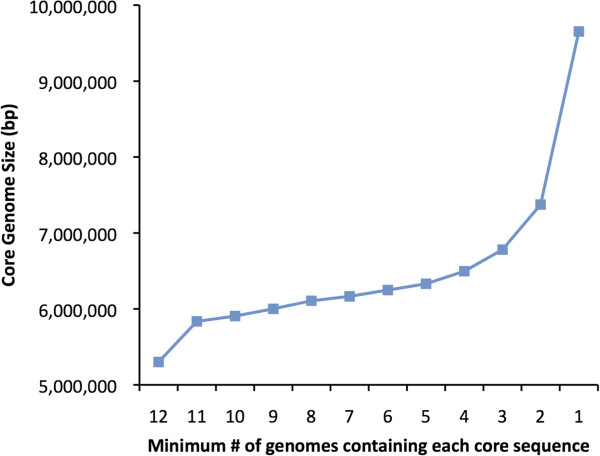
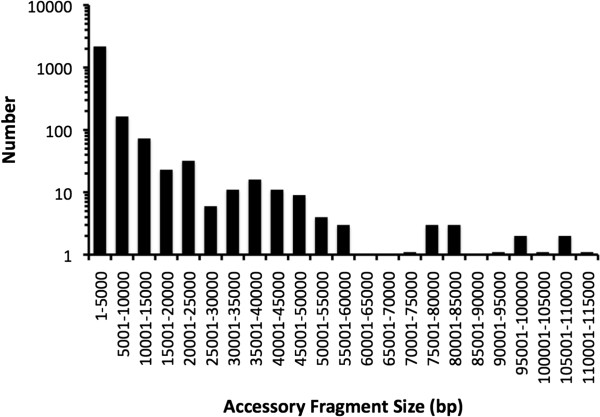
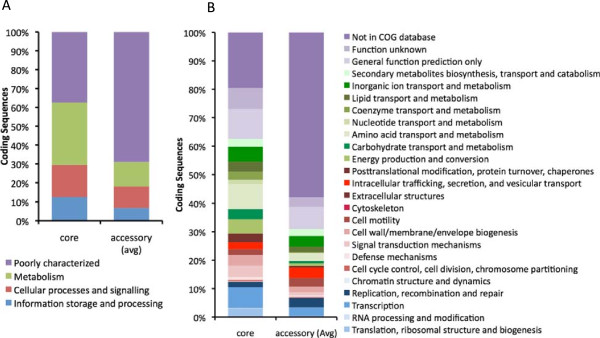
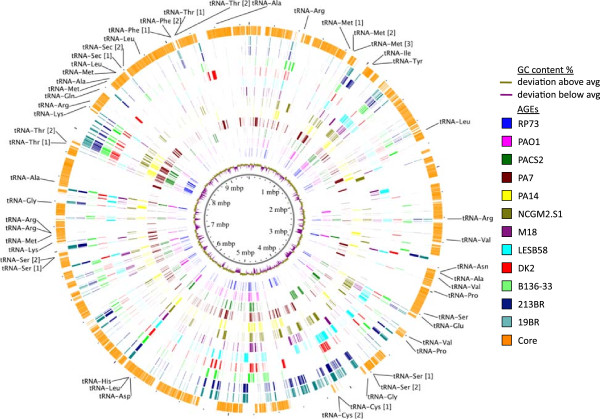
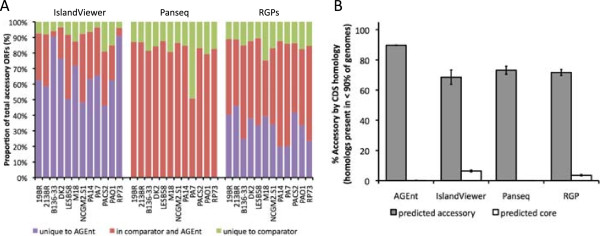
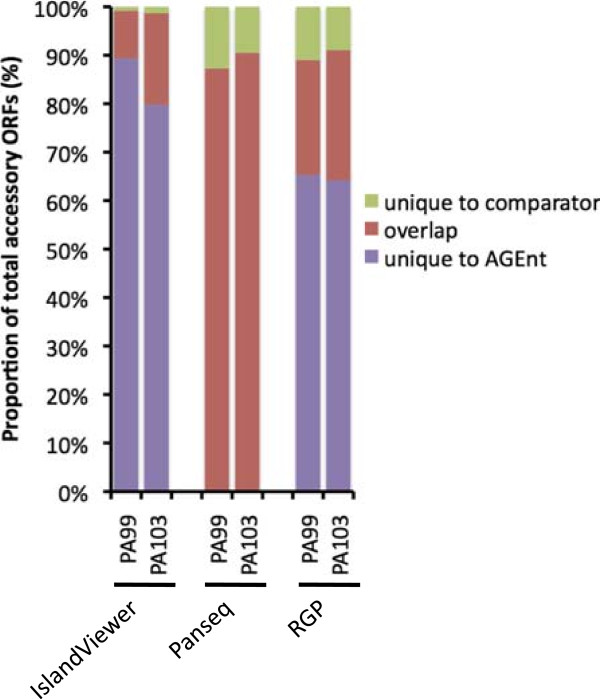
Results: We developed a bioinformatic method for identifying the accessory genome of P. aeruginosa. First, the core genome was determined based on sequence conserved among the completed genomes of twelve reference strains using Spine, a software program developed for this purpose. The core genome was 5.84 Mbp in size and contained 5,316 coding sequences. We then developed an in silico genome subtraction program named AGEnt to filter out core genomic sequences from P. aeruginosa whole genomes to identify accessory genomic sequences of these reference strains. This analysis determined that the accessory genome of P. aeruginosa ranged from 6.9-18.0% of the total genome, was enriched for genes associated with mobile elements, and was comprised of a majority of genes with unknown or unclear function. Using these genomes, we showed that AGEnt performed well compared to other publically available programs designed to detect accessory genomic elements. We then demonstrated the utility of the AGEnt program by applying it to the draft genomes of two previously unsequenced P. aeruginosa strains, PA99 and PA103.
Conclusions: The P. aeruginosa genome is rich in accessory genetic material. The AGEnt program accurately identified the accessory genomes of newly sequenced P. aeruginosa strains, even when draft genomes were used. As P. aeruginosa genomes become available at an increasingly rapid pace, this program will be useful in cataloging the expanding accessory genome of this bacterium and in discerning correlations between phenotype and accessory genome makeup. The combination of Spine and AGEnt should be useful in defining the accessory genomes of other bacterial species as well.
Figures
References
-
- Jarvis WR. Epidemiology and Control of Pseudomonas aeruginosa Infections in the Intensive Care Unit. In: Hauser AR, Rello J, editors. Severe Infections Caused by Pseudomonas aeruginosa. Boston: Kluwer Academic Publishers; 2003. pp. 153–168.
-
- Obritsch MD, Fish DN, MacLaren R, Jung R. National surveillance of antimicrobial resistance in Pseudomonas aeruginosa isolates obtained from intensive care unit patients from 1993 to 2002. Antimicrob Agents Chemother. 2004;48(12):4606–4610. doi: 10.1128/AAC.48.12.4606-4610.2004. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
- AI04831/AI/NIAID NIH HHS/United States
- R01 AI075191/AI/NIAID NIH HHS/United States
- K24 AI104831/AI/NIAID NIH HHS/United States
- F32 AI089068/AI/NIAID NIH HHS/United States
- AI075191/AI/NIAID NIH HHS/United States
- T32 AI007476/AI/NIAID NIH HHS/United States
- AI053674/AI/NIAID NIH HHS/United States
- R01 AI053674/AI/NIAID NIH HHS/United States
- T32AI007476/AI/NIAID NIH HHS/United States
- R21 AI088286/AI/NIAID NIH HHS/United States
- F32AI108247-01/AI/NIAID NIH HHS/United States
- AI099269/AI/NIAID NIH HHS/United States
- F32 AI108247/AI/NIAID NIH HHS/United States
- AI088286/AI/NIAID NIH HHS/United States
- F32AI089068/AI/NIAID NIH HHS/United States
- R01 AI099269/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
