

Impaired function is a common feature of neuropathy-associated glycyl-tRNA synthetase mutations
- PMID: 25168514
- PMCID: PMC4213347
- DOI: 10.1002/humu.22681
Impaired function is a common feature of neuropathy-associated glycyl-tRNA synthetase mutations
Abstract
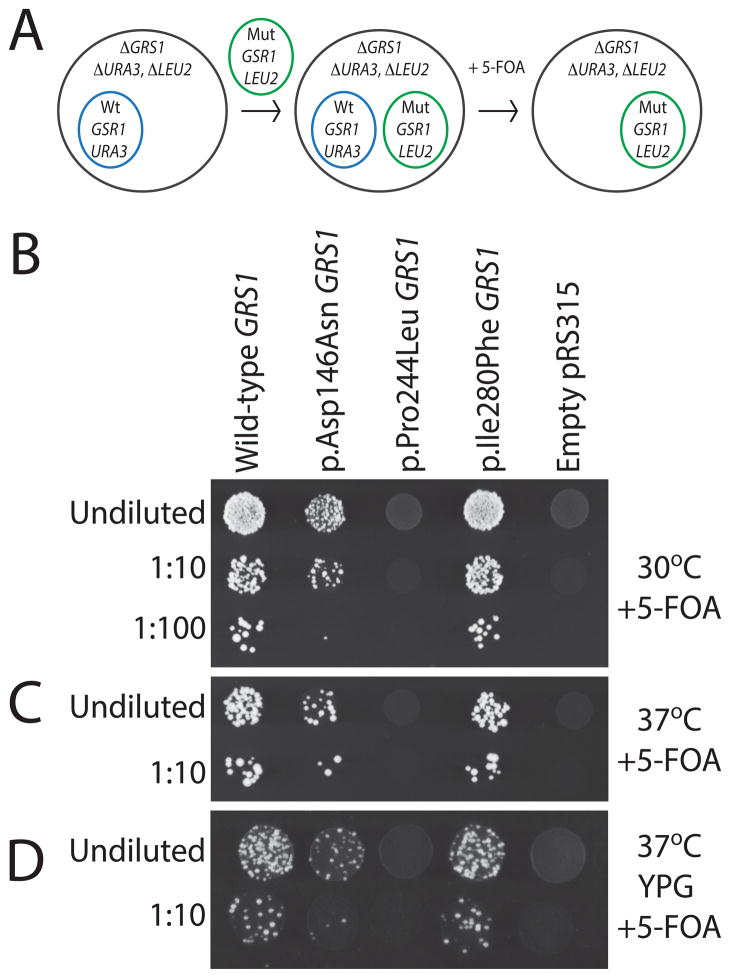
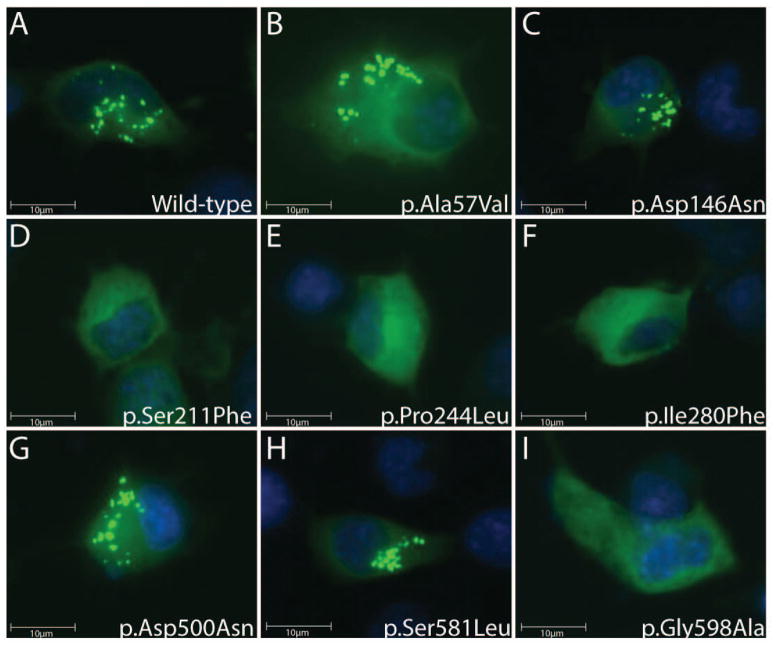
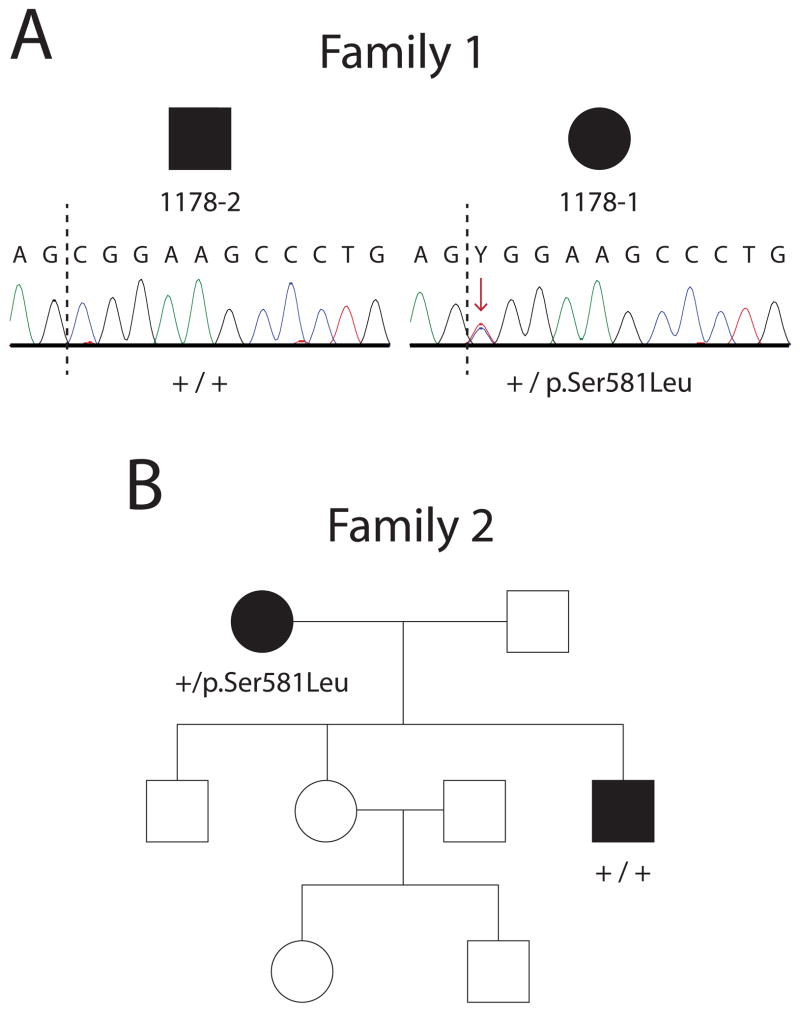
Charcot-Marie-Tooth disease type 2D (CMT2D) is an autosomal-dominant axonal peripheral neuropathy characterized by impaired motor and sensory function in the distal extremities. Mutations in the glycyl-tRNA synthetase (GARS) gene cause CMT2D. GARS is a member of the ubiquitously expressed aminoacyl-tRNA synthetase (ARS) family and is responsible for charging tRNA with glycine. To date, 13 GARS mutations have been identified in patients with CMT disease. While functional studies have revealed loss-of-function characteristics, only four GARS mutations have been rigorously studied. Here, we report the functional evaluation of nine CMT-associated GARS mutations in tRNA charging, yeast complementation, and subcellular localization assays. Our results demonstrate that impaired function is a common characteristic of CMT-associated GARS mutations. Additionally, one mutation previously associated with CMT disease (p.Ser581Leu) does not demonstrate impaired function, was identified in the general population, and failed to segregate with disease in two newly identified families with CMT disease. Thus, we propose that this variant is not a disease-causing mutation. Together, our data indicate that impaired function is a key component of GARS-mediated CMT disease and emphasize the need for careful genetic and functional evaluation before implicating a variant in disease onset.
Keywords: Charcot-Marie-Tooth disease; GARS; aminoacyl-tRNA synthetase; glycyl-tRNA synthetase; peripheral neuropathy.
© 2014 WILEY PERIODICALS, INC.
Figures

References
-
- Abe A, Hayasaka K. The GARS gene is rarely mutated in Japanese patients with Charcot-Marie-Tooth neuropathy. Journal of human genetics. 2009;54:310–312. - PubMed
-
- Achilli F, Bros-Facer V, Williams HP, Banks GT, AlQatari M, Chia R, Tucci V, Groves M, Nickols CD, Seburn KL, Kendall R, Cader MZ, et al. An ENU-induced mutation in mouse glycyl-tRNA synthetase (GARS) causes peripheral sensory and motor phenotypes creating a model of Charcot-Marie-Tooth type 2D peripheral neuropathy. Disease Models & Mechanisms. 2009;2:359–373. - PMC - PubMed
-
- Antonellis A, Ellsworth RE, Sambuughin N, Puls I, Abel A, Lee-Lin S-Q, Jordanova A, Kremensky I, Christodoulou K, Middleton LT, Sivakumar K, Ionasescu V, et al. Glycyl tRNA Synthetase Mutations in Charcot-Marie-Tooth Disease Type 2D and Distal Spinal Muscular Atrophy Type V. The American Journal of Human Genetics. 2003;72:1293–1299. - PMC - PubMed
-
- Antonellis A, Green ED. The Role of Aminoacyl-tRNA Synthetases in Genetic Diseases*. Annual Review of Genomics and Human Genetics. 2008;9:87–107. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
