

The architecture of a scrambled genome reveals massive levels of genomic rearrangement during development
- PMID: 25171416
- PMCID: PMC4199391
- DOI: 10.1016/j.cell.2014.07.034
The architecture of a scrambled genome reveals massive levels of genomic rearrangement during development
Abstract
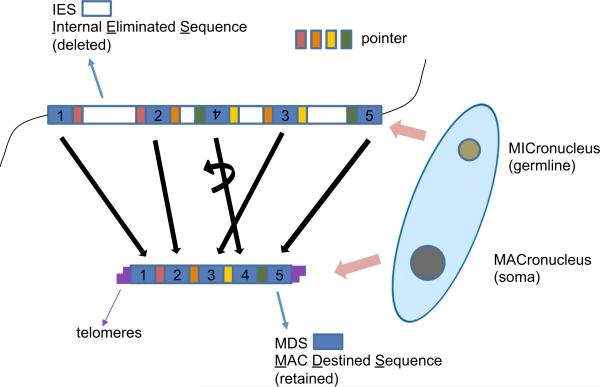
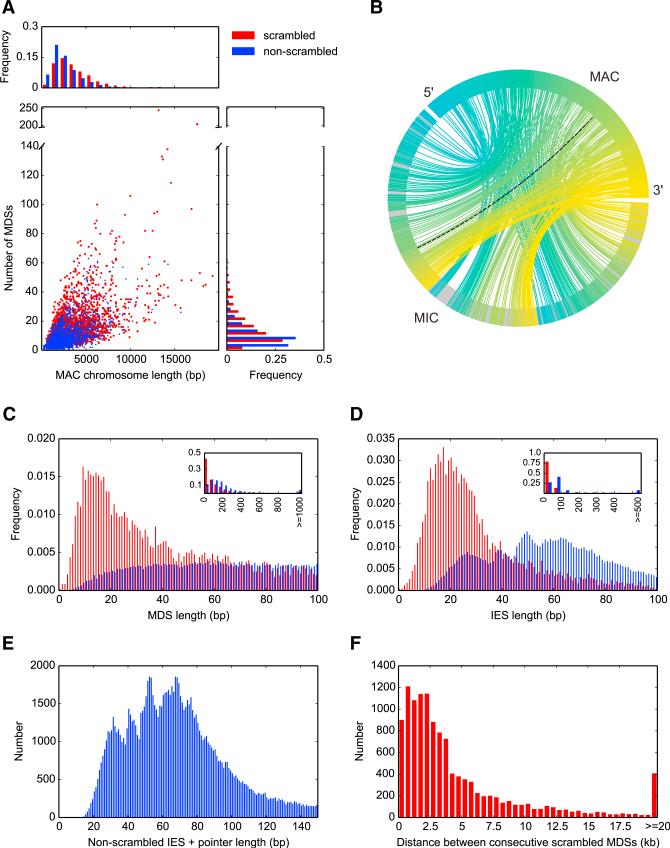
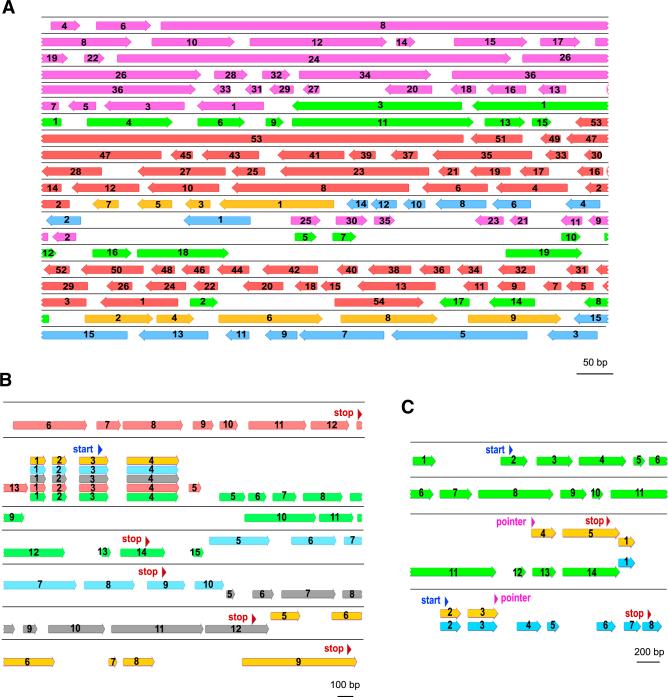
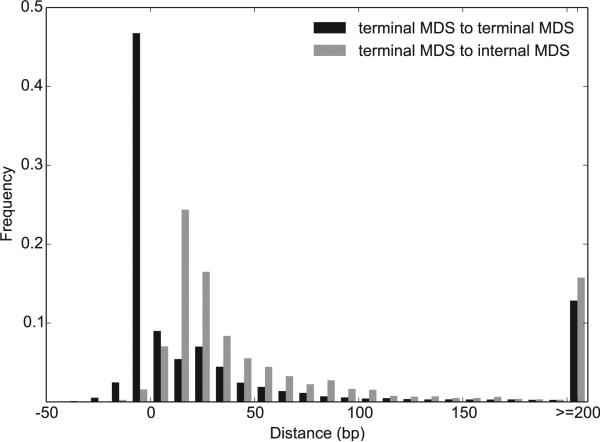
Programmed DNA rearrangements in the single-celled eukaryote Oxytricha trifallax completely rewire its germline into a somatic nucleus during development. This elaborate, RNA-mediated pathway eliminates noncoding DNA sequences that interrupt gene loci and reorganizes the remaining fragments by inversions and permutations to produce functional genes. Here, we report the Oxytricha germline genome and compare it to the somatic genome to present a global view of its massive scale of genome rearrangements. The remarkably encrypted genome architecture contains >3,500 scrambled genes, as well as >800 predicted germline-limited genes expressed, and some posttranslationally modified, during genome rearrangements. Gene segments for different somatic loci often interweave with each other. Single gene segments can contribute to multiple, distinct somatic loci. Terminal precursor segments from neighboring somatic loci map extremely close to each other, often overlapping. This genome assembly provides a draft of a scrambled genome and a powerful model for studies of genome rearrangement.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures
References
-
- Aury J-M, Jaillon O, Duret L, Noel B, Jubin C, Porcel BM, Ségurens B, Daubin V, Anthouard V, Aiach N, et al. Global trends of whole-genome duplications revealed by the ciliate Paramecium tetraurelia. Nature. 2006;444:171–178. - PubMed
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
