

Ecology and evolution of viruses infecting uncultivated SUP05 bacteria as revealed by single-cell- and meta-genomics
- PMID: 25171894
- PMCID: PMC4164917
- DOI: 10.7554/eLife.03125
Ecology and evolution of viruses infecting uncultivated SUP05 bacteria as revealed by single-cell- and meta-genomics
Abstract
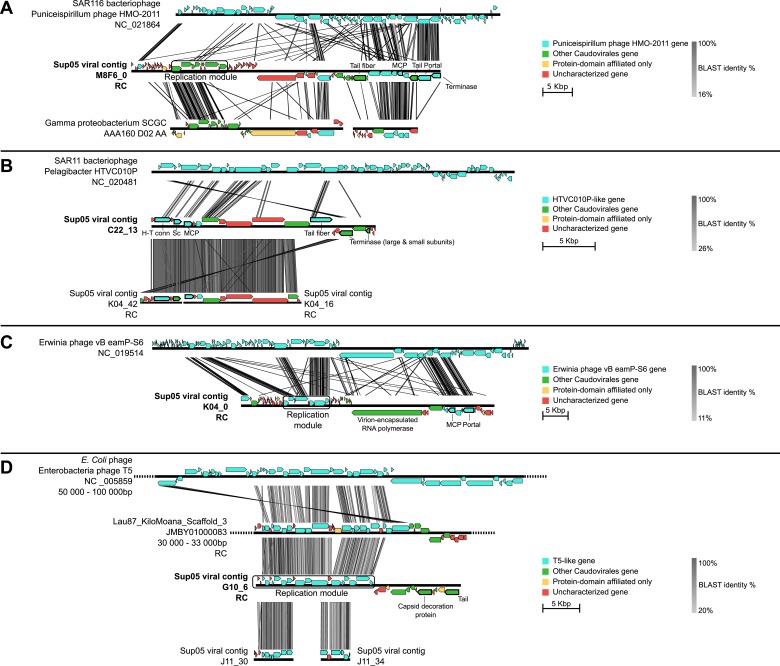
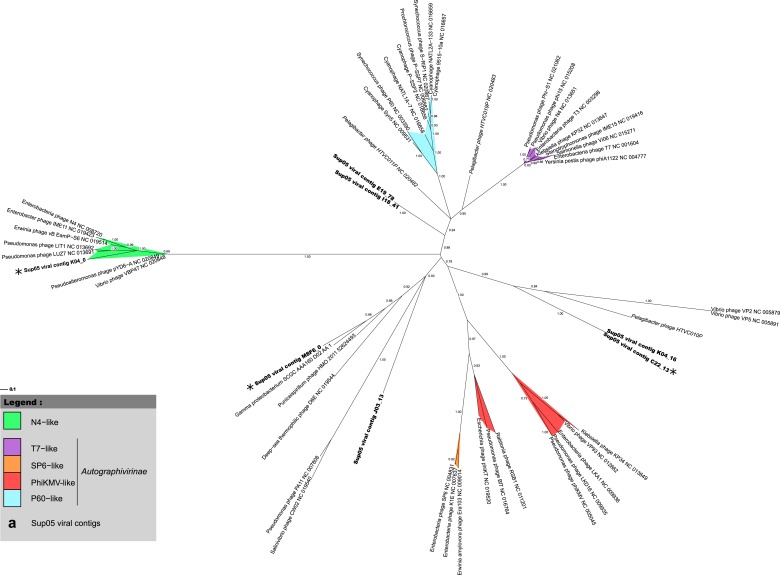
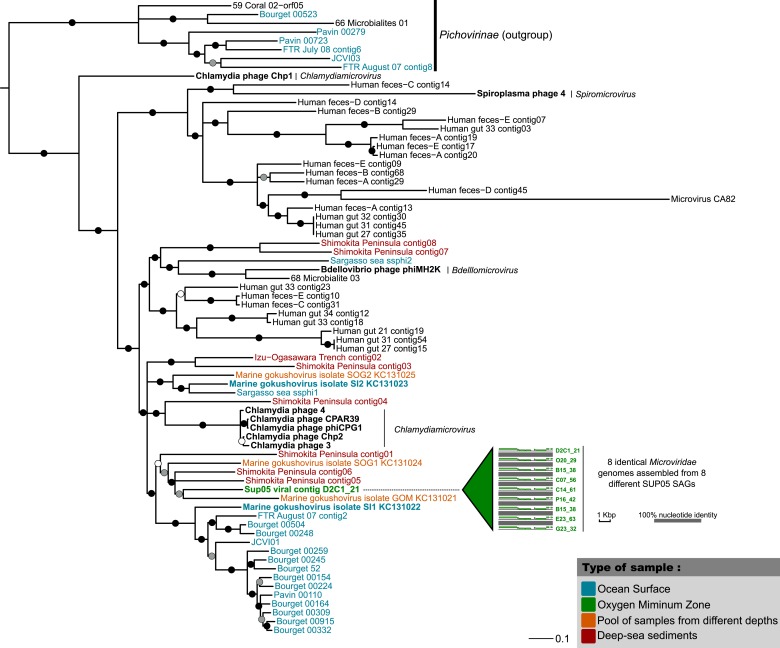
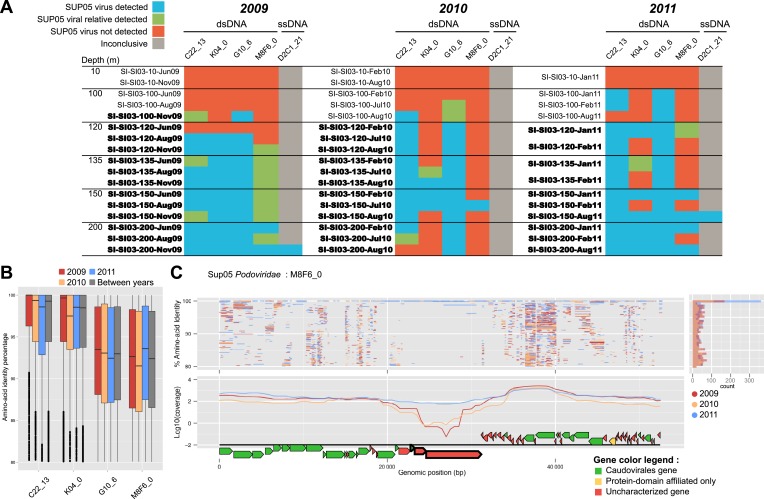
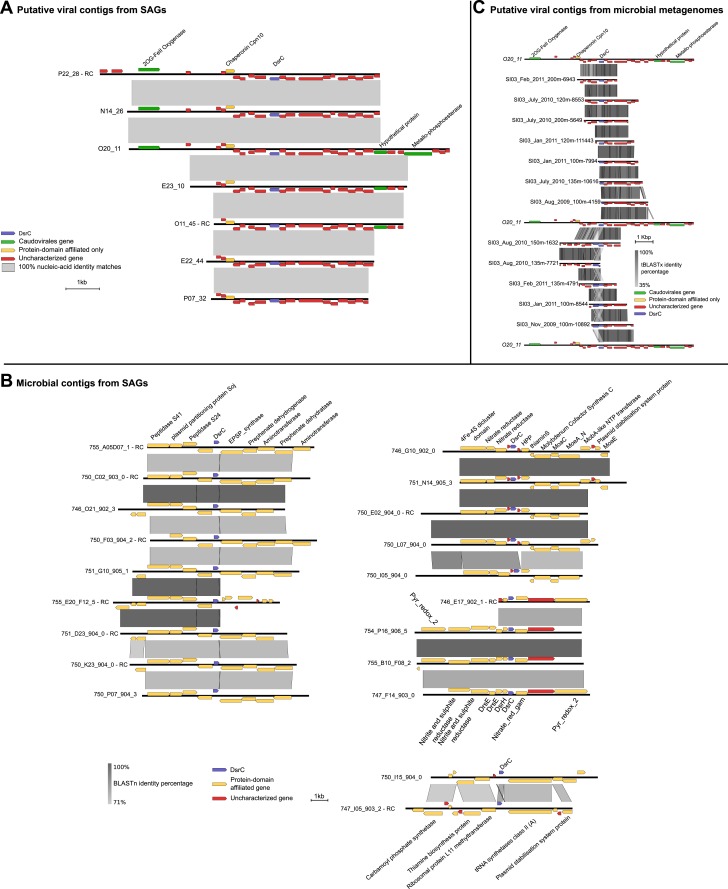
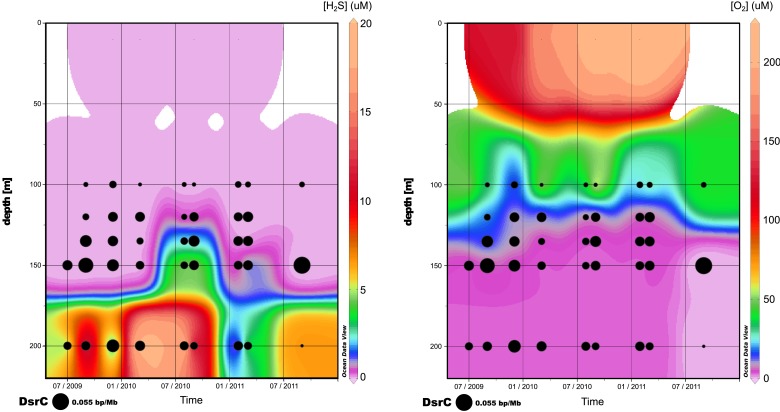
Viruses modulate microbial communities and alter ecosystem functions. However, due to cultivation bottlenecks, specific virus-host interaction dynamics remain cryptic. In this study, we examined 127 single-cell amplified genomes (SAGs) from uncultivated SUP05 bacteria isolated from a model marine oxygen minimum zone (OMZ) to identify 69 viral contigs representing five new genera within dsDNA Caudovirales and ssDNA Microviridae. Infection frequencies suggest that ∼1/3 of SUP05 bacteria is viral-infected, with higher infection frequency where oxygen-deficiency was most severe. Observed Microviridae clonality suggests recovery of bloom-terminating viruses, while systematic co-infection between dsDNA and ssDNA viruses posits previously unrecognized cooperation modes. Analyses of 186 microbial and viral metagenomes revealed that SUP05 viruses persisted for years, but remained endemic to the OMZ. Finally, identification of virus-encoded dissimilatory sulfite reductase suggests SUP05 viruses reprogram their host's energy metabolism. Together, these results demonstrate closely coupled SUP05 virus-host co-evolutionary dynamics with the potential to modulate biogeochemical cycling in climate-critical and expanding OMZs.
Keywords: SUP05; bacteriophages; ecology; infectious disease; microbiology; oxygen minimum zone; single cell genomics; viral dark matter; viruses.
Conflict of interest statement
The authors declare that no competing interests exist.
Figures
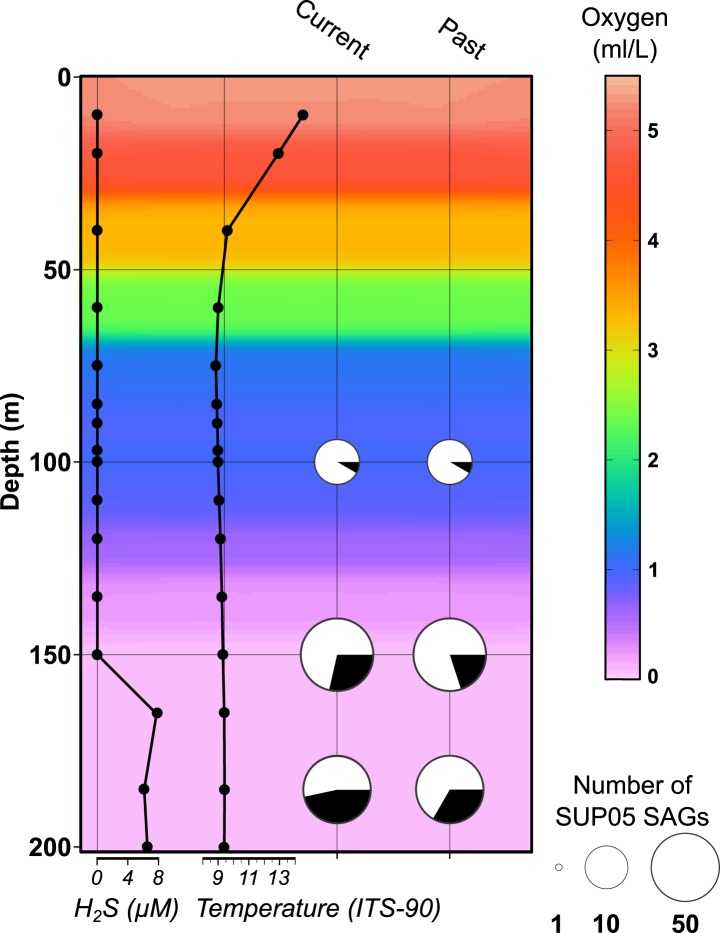
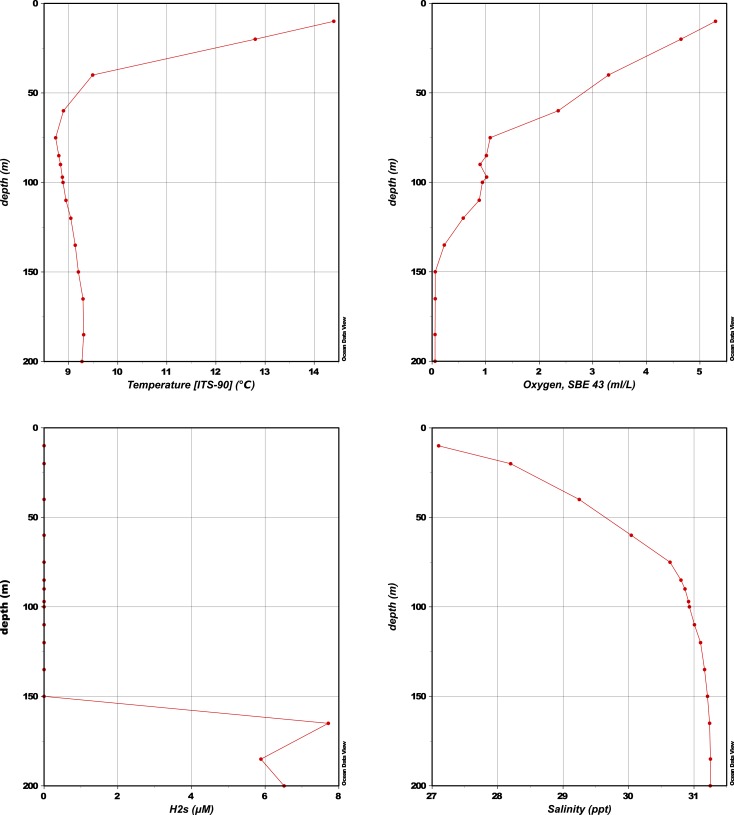
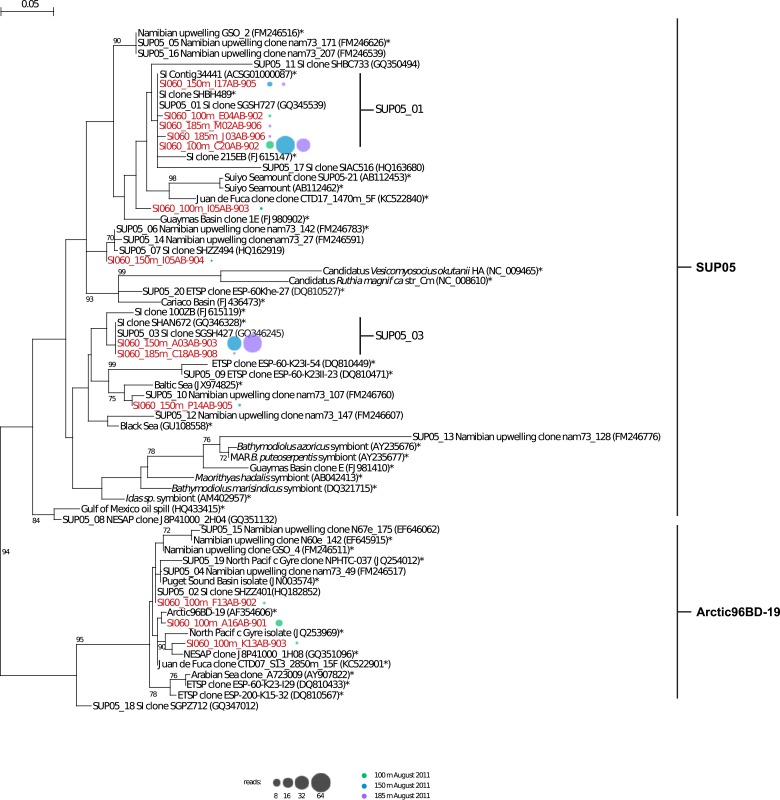
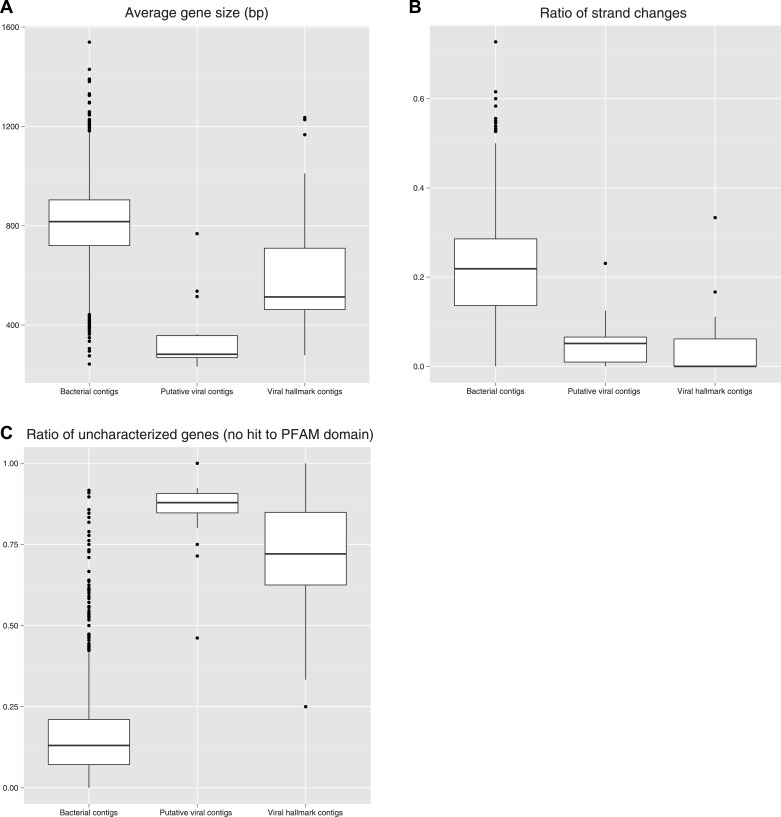

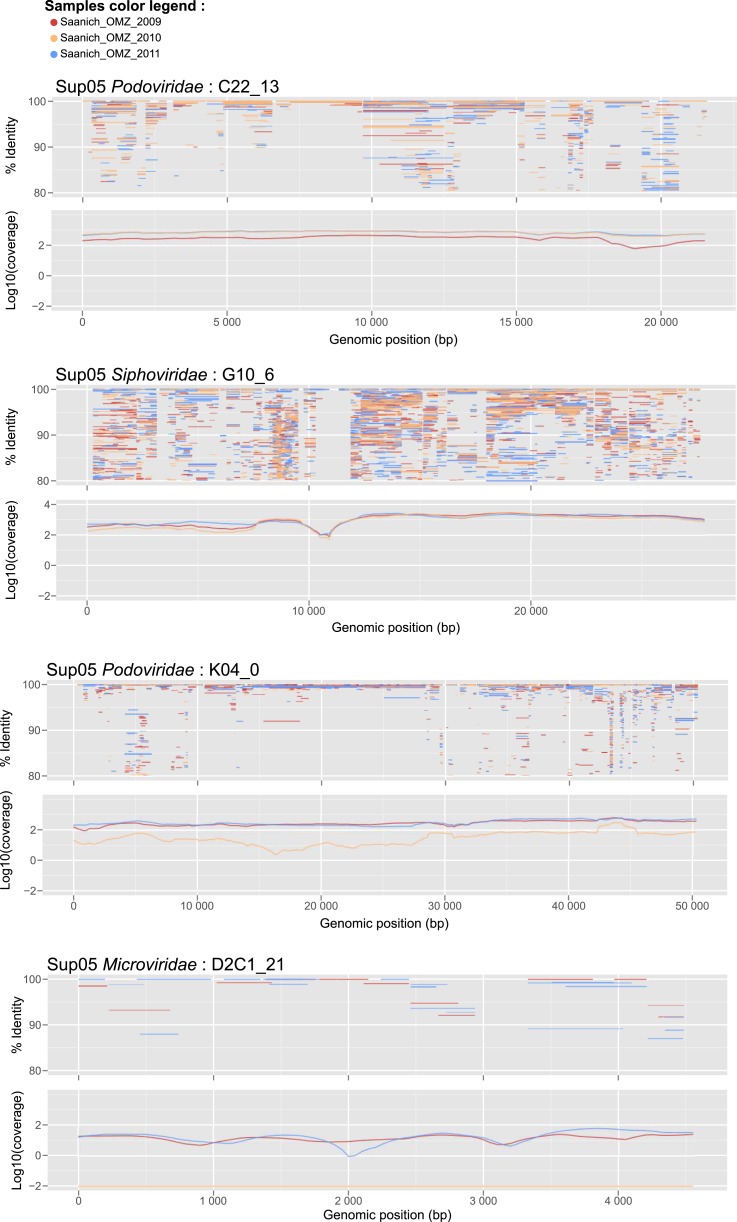
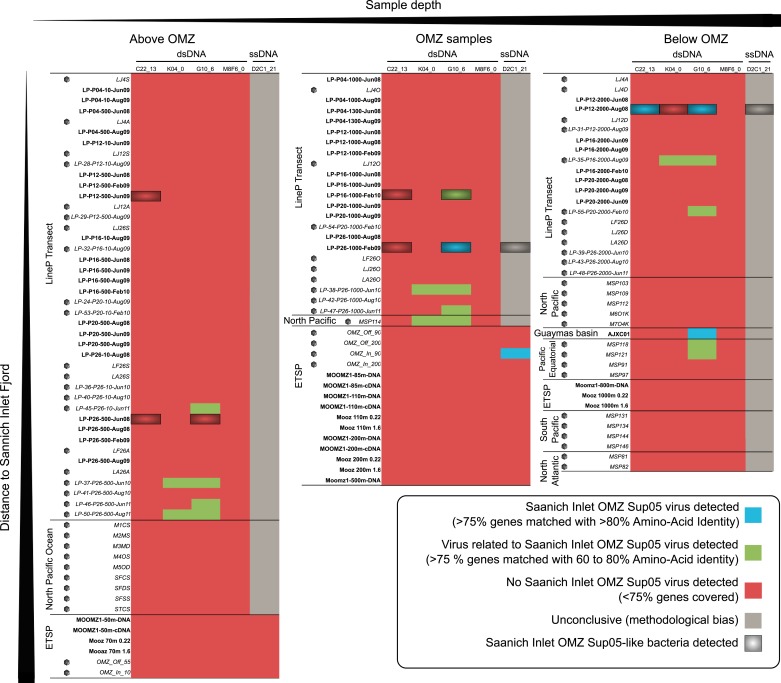
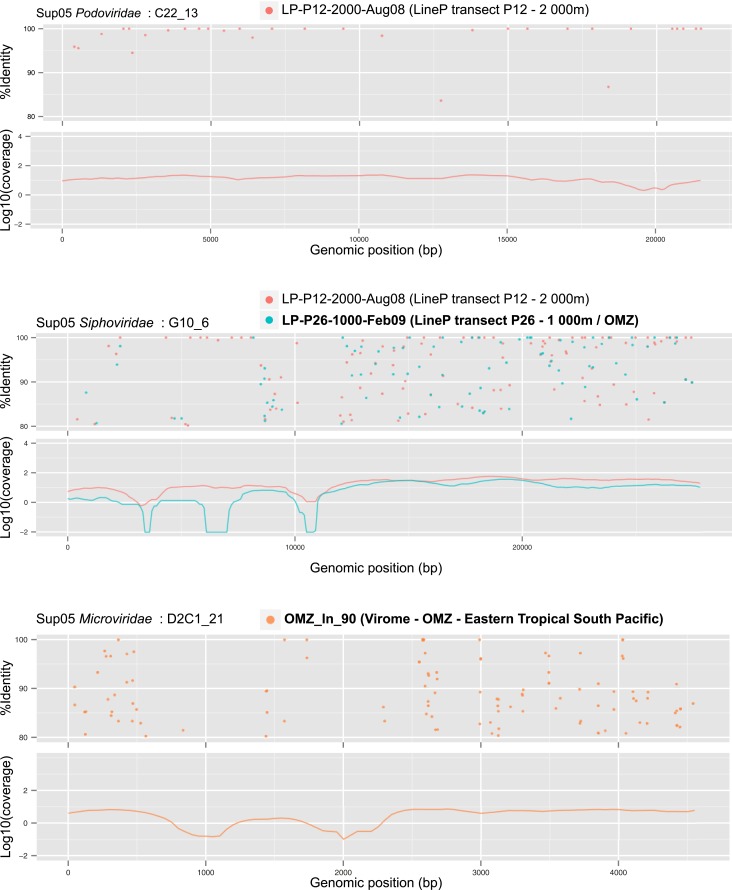
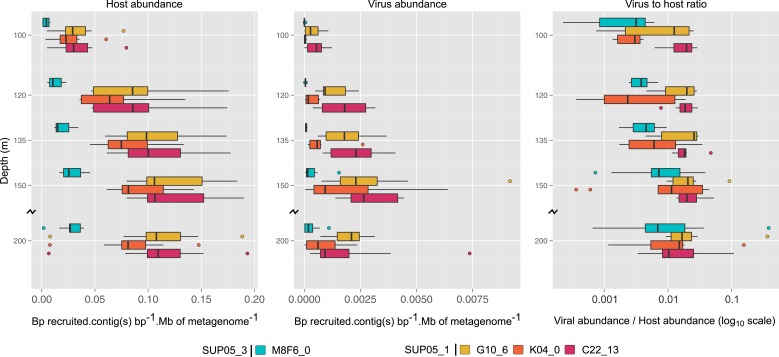

Comment in
-
Gene swapping in the dead zone.Elife. 2014 Oct 13;3:e04600. doi: 10.7554/eLife.04600. Elife. 2014. PMID: 25310237 Free PMC article.
Similar articles
-
Gene swapping in the dead zone.Elife. 2014 Oct 13;3:e04600. doi: 10.7554/eLife.04600. Elife. 2014. PMID: 25310237 Free PMC article.
-
Metagenome of a versatile chemolithoautotroph from expanding oceanic dead zones.Science. 2009 Oct 23;326(5952):578-82. doi: 10.1126/science.1175309. Science. 2009. PMID: 19900896
-
Oxygen minimum zone cryptic sulfur cycling sustained by offshore transport of key sulfur oxidizing bacteria.Nat Commun. 2018 Apr 30;9(1):1729. doi: 10.1038/s41467-018-04041-x. Nat Commun. 2018. PMID: 29712903 Free PMC article.
-
Single-stranded DNA viruses employ a variety of mechanisms for integration into host genomes.Ann N Y Acad Sci. 2015 Apr;1341:41-53. doi: 10.1111/nyas.12675. Epub 2015 Feb 11. Ann N Y Acad Sci. 2015. PMID: 25675979 Review.
-
The bacterial sulfur cycle in expanding dysoxic and euxinic marine waters.Environ Microbiol. 2021 Jun;23(6):2834-2857. doi: 10.1111/1462-2920.15265. Epub 2020 Oct 18. Environ Microbiol. 2021. PMID: 33000514 Free PMC article. Review.
Cited by
-
Limitations of Correlation-Based Inference in Complex Virus-Microbe Communities.mSystems. 2018 Aug 28;3(4):e00084-18. doi: 10.1128/mSystems.00084-18. eCollection 2018 Jul-Aug. mSystems. 2018. PMID: 30175237 Free PMC article.
-
Microbial Ecology of Oxygen Minimum Zones Amidst Ocean Deoxygenation.Front Microbiol. 2021 Oct 27;12:748961. doi: 10.3389/fmicb.2021.748961. eCollection 2021. Front Microbiol. 2021. PMID: 34777296 Free PMC article. Review.
-
Phage diversity, genomics and phylogeny.Nat Rev Microbiol. 2020 Mar;18(3):125-138. doi: 10.1038/s41579-019-0311-5. Epub 2020 Feb 3. Nat Rev Microbiol. 2020. PMID: 32015529 Review.
-
Archaeal Viruses from High-Temperature Environments.Genes (Basel). 2018 Feb 27;9(3):128. doi: 10.3390/genes9030128. Genes (Basel). 2018. PMID: 29495485 Free PMC article. Review.
-
Delta-proteobacterial SAR324 group in hydrothermal plumes on the South Mid-Atlantic Ridge.Sci Rep. 2016 Mar 8;6:22842. doi: 10.1038/srep22842. Sci Rep. 2016. PMID: 26953077 Free PMC article.
References
-
- Allers E, Moraru C, Duhaime MB, Beneze E, Solonenko N, Canosa JB, Amann R, Sullivan MB. Single-cell and population level viral infection dynamics revealed by phageFISH, a method to visualize intracellular and free viruses. Environmental Microbiology. 2013;15:2306–2318. doi: 10.1111/1462-2920.12100. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
