

Evidence for deep regulatory similarities in early developmental programs across highly diverged insects
- PMID: 25173756
- PMCID: PMC4217690
- DOI: 10.1093/gbe/evu184
Evidence for deep regulatory similarities in early developmental programs across highly diverged insects
Abstract
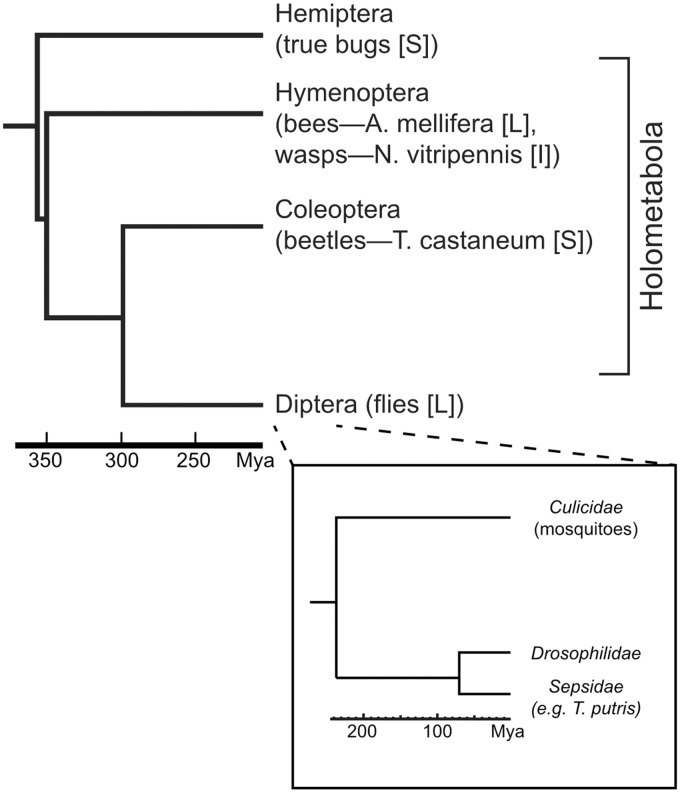
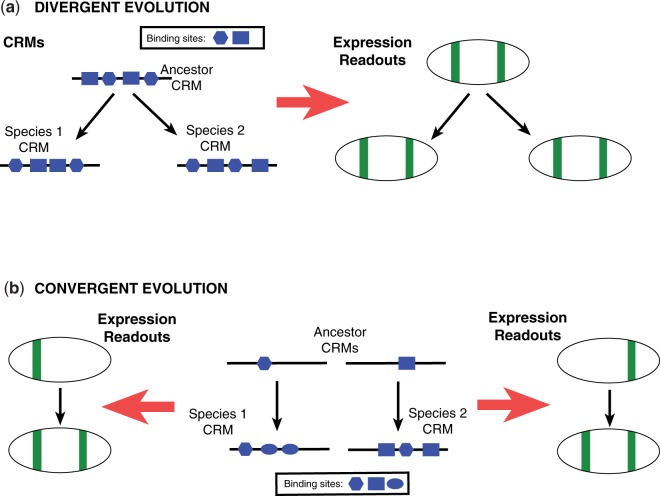
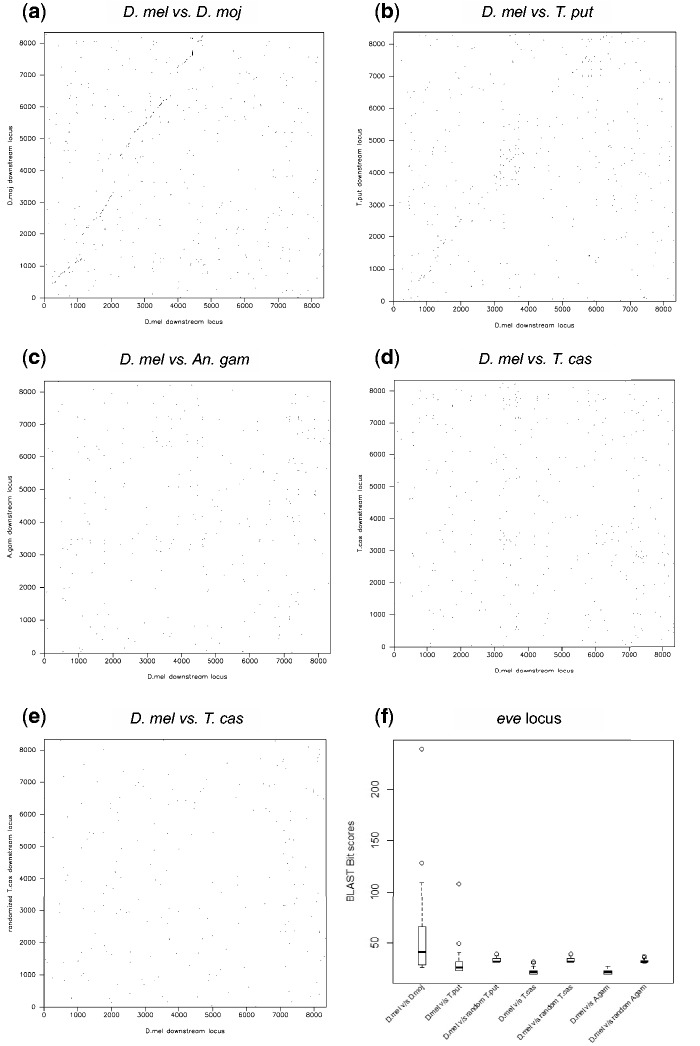
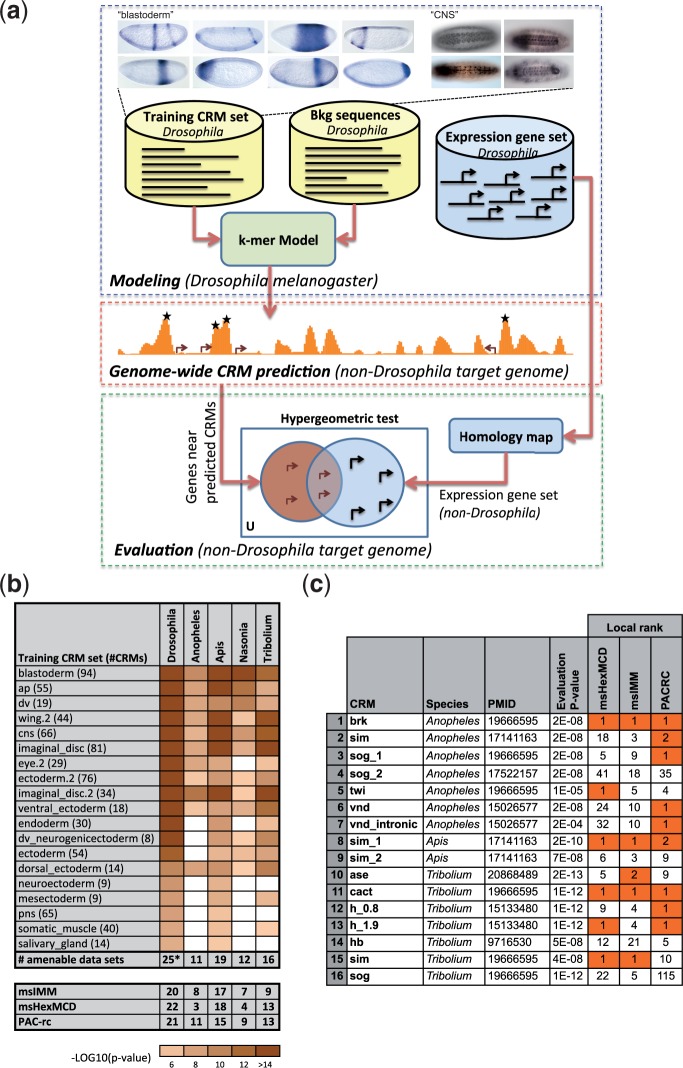
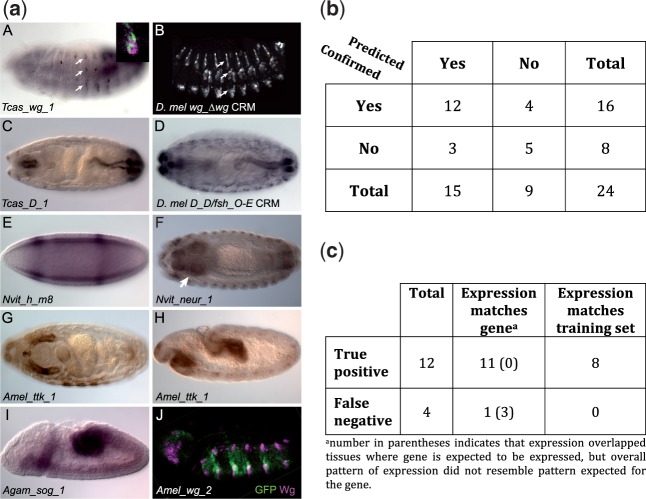
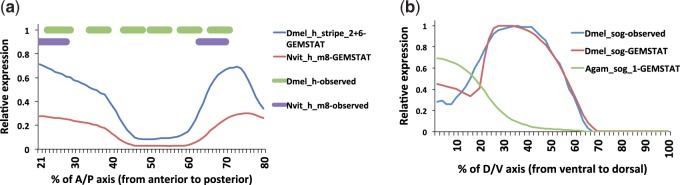
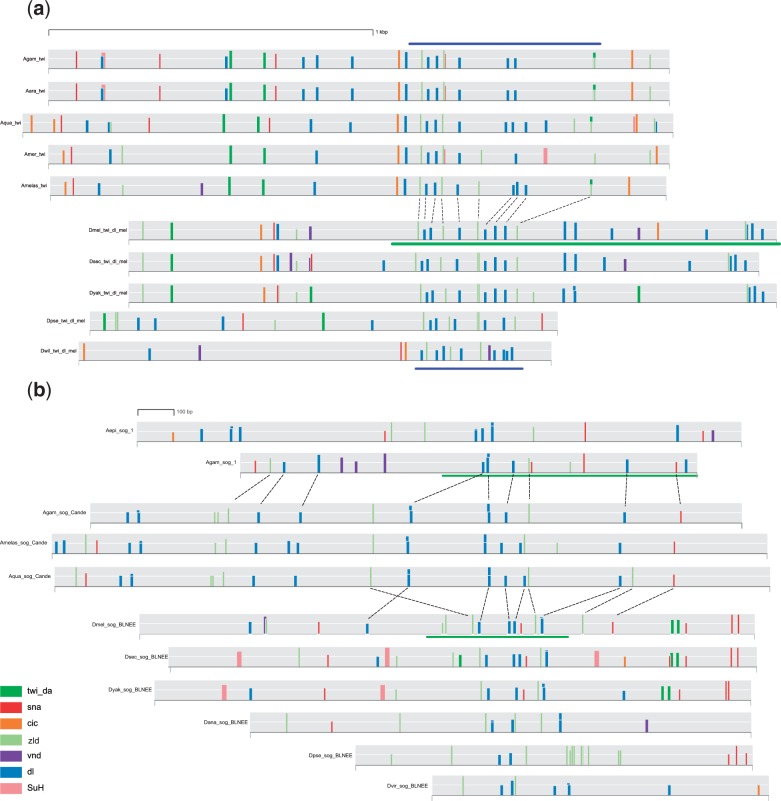
Many genes familiar from Drosophila development, such as the so-called gap, pair-rule, and segment polarity genes, play important roles in the development of other insects and in many cases appear to be deployed in a similar fashion, despite the fact that Drosophila-like "long germband" development is highly derived and confined to a subset of insect families. Whether or not these similarities extend to the regulatory level is unknown. Identification of regulatory regions beyond the well-studied Drosophila has been challenging as even within the Diptera (flies, including mosquitoes) regulatory sequences have diverged past the point of recognition by standard alignment methods. Here, we demonstrate that methods we previously developed for computational cis-regulatory module (CRM) discovery in Drosophila can be used effectively in highly diverged (250-350 Myr) insect species including Anopheles gambiae, Tribolium castaneum, Apis mellifera, and Nasonia vitripennis. In Drosophila, we have successfully used small sets of known CRMs as "training data" to guide the search for other CRMs with related function. We show here that although species-specific CRM training data do not exist, training sets from Drosophila can facilitate CRM discovery in diverged insects. We validate in vivo over a dozen new CRMs, roughly doubling the number of known CRMs in the four non-Drosophila species. Given the growing wealth of Drosophila CRM annotation, these results suggest that extensive regulatory sequence annotation will be possible in newly sequenced insects without recourse to costly and labor-intensive genome-scale experiments. We develop a new method, Regulus, which computes a probabilistic score of similarity based on binding site composition (despite the absence of nucleotide-level sequence alignment), and demonstrate similarity between functionally related CRMs from orthologous loci. Our work represents an important step toward being able to trace the evolutionary history of gene regulatory networks and defining the mechanisms underlying insect evolution.
Keywords: alignment free enhancer prediction; cross species enhancer discovery; regulatory modules in diverged insects.
© The Author(s) 2014. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures

Similar articles
-
CRM Discovery Beyond Model Insects.Methods Mol Biol. 2019;1858:117-139. doi: 10.1007/978-1-4939-8775-7_10. Methods Mol Biol. 2019. PMID: 30414115 Free PMC article.
-
Functional evolution of cis-regulatory modules at a homeotic gene in Drosophila.PLoS Genet. 2009 Nov;5(11):e1000709. doi: 10.1371/journal.pgen.1000709. Epub 2009 Nov 6. PLoS Genet. 2009. PMID: 19893611 Free PMC article.
-
RNA editing and alternative splicing of the insect nAChR subunit alpha6 transcript: evolutionary conservation, divergence and regulation.BMC Evol Biol. 2007 Jun 27;7:98. doi: 10.1186/1471-2148-7-98. BMC Evol Biol. 2007. PMID: 17597521 Free PMC article.
-
Neuropeptides in the nervous system of Drosophila and other insects: multiple roles as neuromodulators and neurohormones.Prog Neurobiol. 2002 Sep;68(1):1-84. doi: 10.1016/s0301-0082(02)00057-6. Prog Neurobiol. 2002. PMID: 12427481 Review.
-
A genome-wide inventory of neurohormone GPCRs in the red flour beetle Tribolium castaneum.Front Neuroendocrinol. 2008 Jan;29(1):142-65. doi: 10.1016/j.yfrne.2007.10.003. Epub 2007 Oct 24. Front Neuroendocrinol. 2008. PMID: 18054377 Review.
Cited by
-
Enhancer identification and activity evaluation in the red flour beetle, Tribolium castaneum.Development. 2018 Apr 5;145(7):dev160663. doi: 10.1242/dev.160663. Development. 2018. PMID: 29540499 Free PMC article.
-
The Role of Chromatin Accessibility in cis-Regulatory Evolution.Genome Biol Evol. 2019 Jul 1;11(7):1813-1828. doi: 10.1093/gbe/evz103. Genome Biol Evol. 2019. PMID: 31114856 Free PMC article.
-
Behavior-related gene regulatory networks: A new level of organization in the brain.Proc Natl Acad Sci U S A. 2020 Sep 22;117(38):23270-23279. doi: 10.1073/pnas.1921625117. Epub 2020 Jul 13. Proc Natl Acad Sci U S A. 2020. PMID: 32661177 Free PMC article. Review.
-
Comprehensive Genomic Discovery of Non-Coding Transcriptional Enhancers in the African Malaria Vector Anopheles coluzzii.Front Genet. 2022 Jan 10;12:785934. doi: 10.3389/fgene.2021.785934. eCollection 2021. Front Genet. 2022. PMID: 35082832 Free PMC article.
-
SCRMshaw: Supervised cis-regulatory module prediction for insect genomes.PLoS One. 2024 Dec 5;19(12):e0311752. doi: 10.1371/journal.pone.0311752. eCollection 2024. PLoS One. 2024. PMID: 39637210 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
