

Differential diagnosis of usual interstitial pneumonia: when is it truly idiopathic?
- PMID: 25176967
- PMCID: PMC9487316
- DOI: 10.1183/09059180.00004914
Differential diagnosis of usual interstitial pneumonia: when is it truly idiopathic?
Erratum in
- Eur Respir Rev. 2014 Dec 23(134):537
Abstract
Idiopathic pulmonary fibrosis (IPF), the most common and lethal of the idiopathic interstitial pneumonias, is defined by a radiological and/or pathological pattern of usual interstitial pneumonia (UIP). However, UIP is not synonymous with IPF as other clinical conditions may be associated with UIP, including chronic hypersensitivity pneumonitis, collagen vascular disease, drug toxicity, asbestosis, familial IPF and Hermansky-Pudlak syndrome. Differentiating IPF ("idiopathic UIP") from conditions that mimic IPF ("secondary UIP") has substantial therapeutic and prognostic implications. A number of radiological and histological clues may help distinguish IPF from other conditions with a UIP pattern of fibrosis, but their appreciation requires extensive expertise in interstitial lung disease as well as an integrated multidisciplinary approach involving pulmonologists, radiologists and pathologists. In addition, multidisciplinary discussions may decrease the time to initial IPF diagnosis and, thus, enable more timely management. This concept was strongly emphasised by the 2011 ATS/ERS/JRS/ALAT guidelines. This article highlights, with the aid of a clinical case, the difficulties in making a diagnosis of IPF in clinical practice. Yet, an accurate diagnosis is critical, particularly given the availability of drugs that may reduce the pace of functional decline and disease progression in IPF.
©ERS 2014.
Conflict of interest statement
Conflict of interest: Disclosures can be found alongside the online version of this article at
Figures
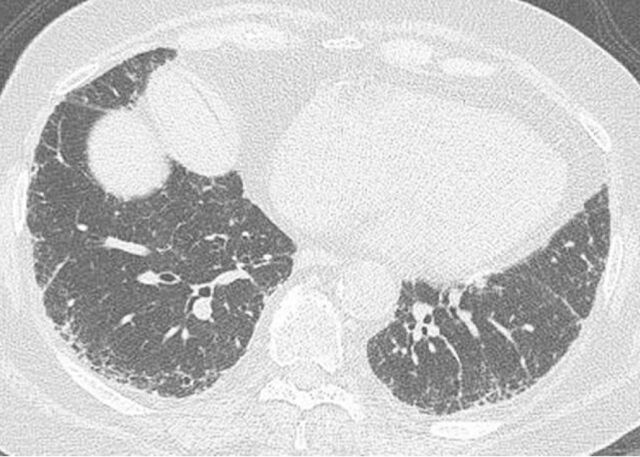
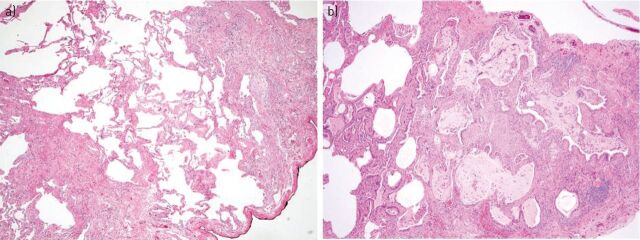
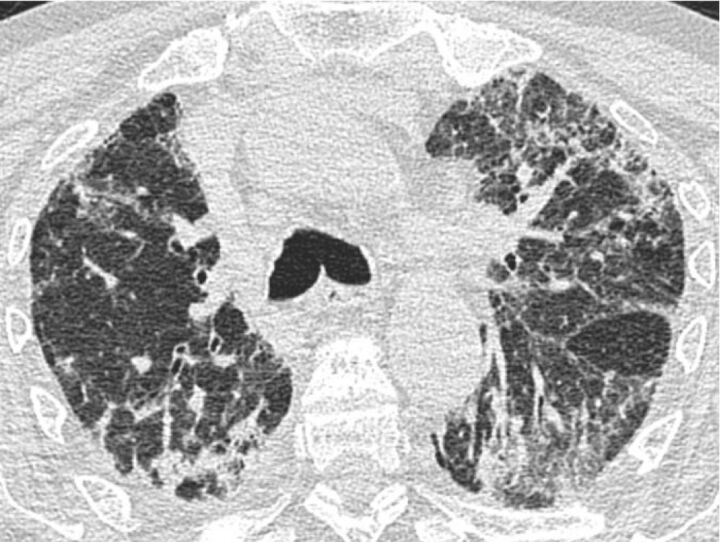

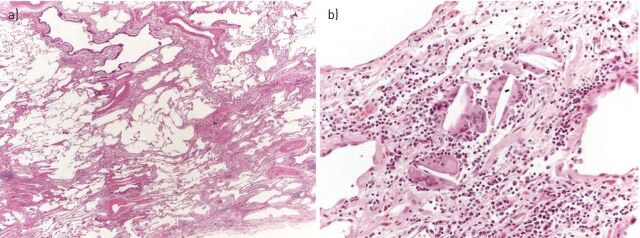
Comment in
-
Inhalation challenge in the differential diagnosis of usual interstitial pneumonia.Eur Respir Rev. 2015 Sep;24(137):542-4. doi: 10.1183/16000617.00000415. Eur Respir Rev. 2015. PMID: 26324817 Free PMC article.
References
-
- Spagnolo P, Du Bois RM, Cottin V. Rare lung disease and orphan drug development. Lancet Respir Med 2013; 1: 479–487. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical