

ALS as a distal axonopathy: molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease
- PMID: 25177267
- PMCID: PMC4132373
- DOI: 10.3389/fnins.2014.00252
ALS as a distal axonopathy: molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease
Abstract
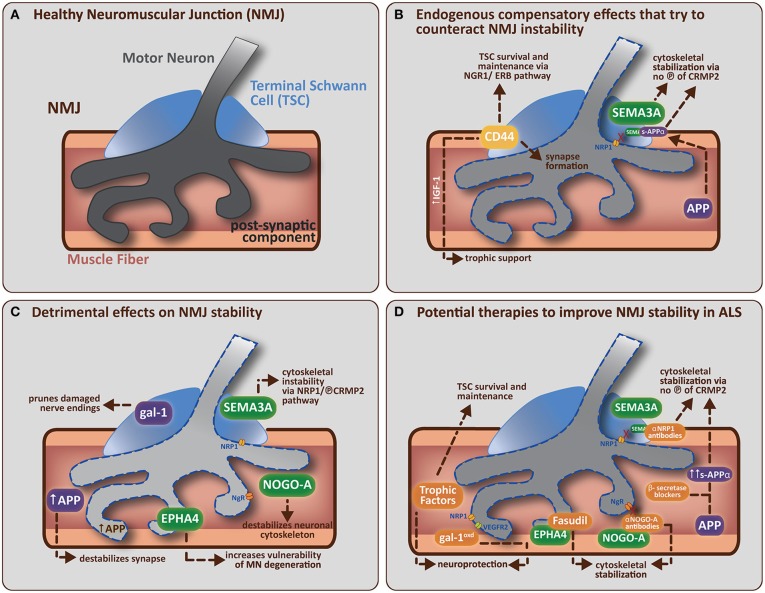
Amyotrophic Lateral Sclerosis (ALS) is being redefined as a distal axonopathy, in that many molecular changes influencing motor neuron degeneration occur at the neuromuscular junction (NMJ) at very early stages of the disease prior to symptom onset. A huge variety of genetic and environmental causes have been associated with ALS, and interestingly, although the cause of the disease can differ, both sporadic and familial forms of ALS show a remarkable similarity in terms of disease progression and clinical manifestation. The NMJ is a highly specialized synapse, allowing for controlled signaling between muscle and nerve necessary for skeletal muscle function. In this review we will evaluate the clinical, animal experimental and cellular/molecular evidence that supports the idea of ALS as a distal axonopathy. We will discuss the early molecular mechanisms that occur at the NMJ, which alter the functional abilities of the NMJ. Specifically, we focus on the role of axon guidance molecules on the stability of the cytoskeleton and how these molecules may directly influence the cells of the NMJ in a way that may initiate or facilitate the dismantling of the neuromuscular synapse in the presymptomatic stages of ALS.
Keywords: Amyotrophic Lateral Sclerosis; axon guidance molecules; distal axonopathy; motor neuron; neuromuscular junction; skeletal muscle; terminal Schwann cell.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous