

Protection after stroke: cellular effectors of neurovascular unit integrity
- PMID: 25177270
- PMCID: PMC4132372
- DOI: 10.3389/fncel.2014.00231
Protection after stroke: cellular effectors of neurovascular unit integrity
Abstract
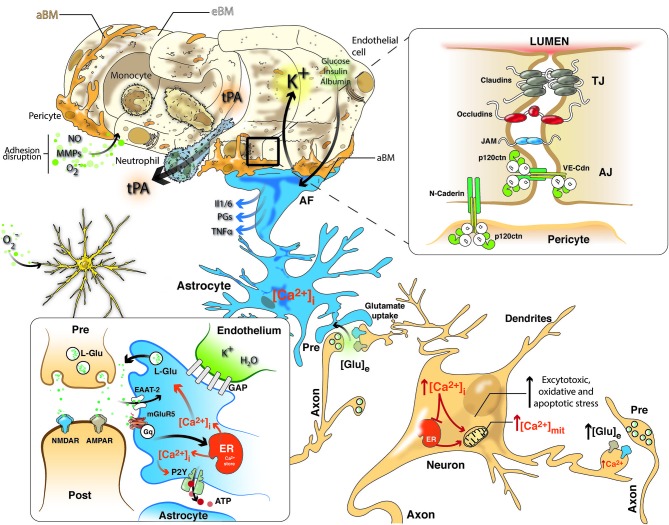
Neurological disorders are prevalent worldwide. Cerebrovascular diseases (CVDs), which account for 55% of all neurological diseases, are the leading cause of permanent disability, cognitive and motor disorders and dementia. Stroke affects the function and structure of blood-brain barrier, the loss of cerebral blood flow regulation, oxidative stress, inflammation and the loss of neural connections. Currently, no gold standard treatments are available outside the acute therapeutic window to improve outcome in stroke patients. Some promising candidate targets have been identified for the improvement of long-term recovery after stroke, such as Rho GTPases, cell adhesion proteins, kinases, and phosphatases. Previous studies by our lab indicated that Rho GTPases (Rac and RhoA) are involved in both tissue damage and survival, as these proteins are essential for the morphology and movement of neurons, astrocytes and endothelial cells, thus playing a critical role in the balance between cell survival and death. Treatment with a pharmacological inhibitor of RhoA/ROCK blocks the activation of the neurodegeneration cascade. In addition, Rac and synaptic adhesion proteins (p120 catenin and N-catenin) play critical roles in protection against cerebral infarction and in recovery by supporting the neurovascular unit and cytoskeletal remodeling activity to maintain the integrity of the brain parenchyma. Interestingly, neuroprotective agents, such as atorvastatin, and CDK5 silencing after cerebral ischemia and in a glutamate-induced excitotoxicity model may act on the same cellular effectors to recover neurovascular unit integrity. Therefore, future efforts must focus on individually targeting the structural and functional roles of each effector of neurovascular unit and the interactions in neural and non-neural cells in the post-ischemic brain and address how to promote the recovery or prevent the loss of homeostasis in the short, medium and long term.
Keywords: BBB; CDK5; NVU; Rho GTPases; p120 catenin; stroke.
Figures

References
-
- Alavi A., Clark C., Fazekas F. (1998). Cerebral ischemia and Alzheimer’s disease: critical role of PET and implications for therapeutic intervention. J. Nucl. Med. 39, 1363–1365 - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous