

Toward a systems-level view of dynamic phosphorylation networks
- PMID: 25177341
- PMCID: PMC4133750
- DOI: 10.3389/fgene.2014.00263
Toward a systems-level view of dynamic phosphorylation networks
Abstract
To better understand how cells sense and respond to their environment, it is important to understand the organization and regulation of the phosphorylation networks that underlie most cellular signal transduction pathways. These networks, which are composed of protein kinases, protein phosphatases and their respective cellular targets, are highly dynamic. Importantly, to achieve signaling specificity, phosphorylation networks must be regulated at several levels, including at the level of protein expression, substrate recognition, and spatiotemporal modulation of enzymatic activity. Here, we briefly summarize some of the traditional methods used to study the phosphorylation status of cellular proteins before focusing our attention on several recent technological advances, such as protein microarrays, quantitative mass spectrometry, and genetically-targetable fluorescent biosensors, that are offering new insights into the organization and regulation of cellular phosphorylation networks. Together, these approaches promise to lead to a systems-level view of dynamic phosphorylation networks.
Keywords: cell signaling and regulation; fluorescent biosensors; kinase-substrate relationship; phosphoproteomics; protein microarrays; protein phosphorylation networks; quantitative mass spectrometry; systems biology.
Figures
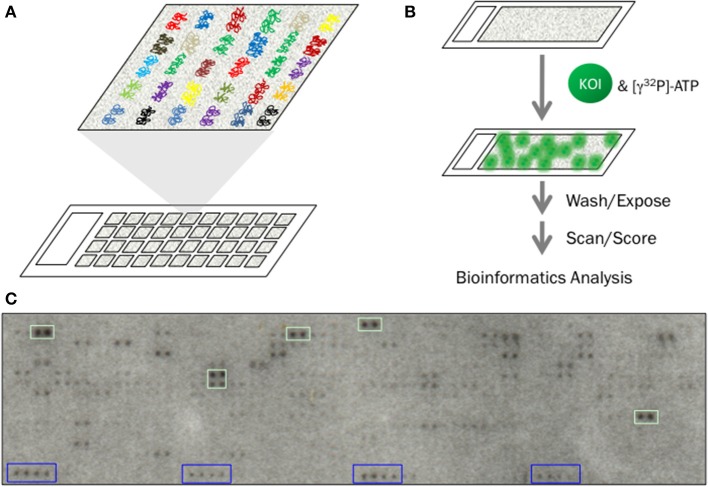
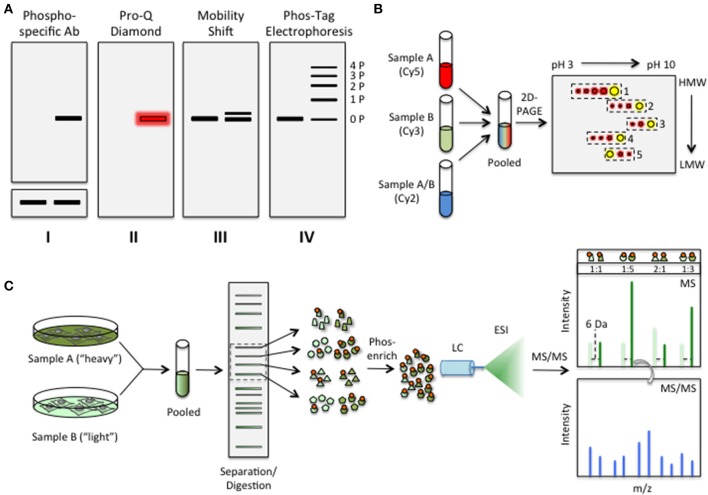
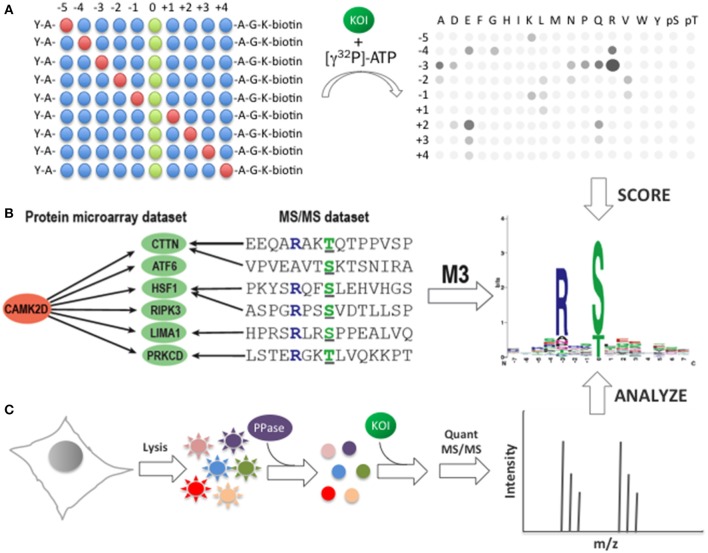
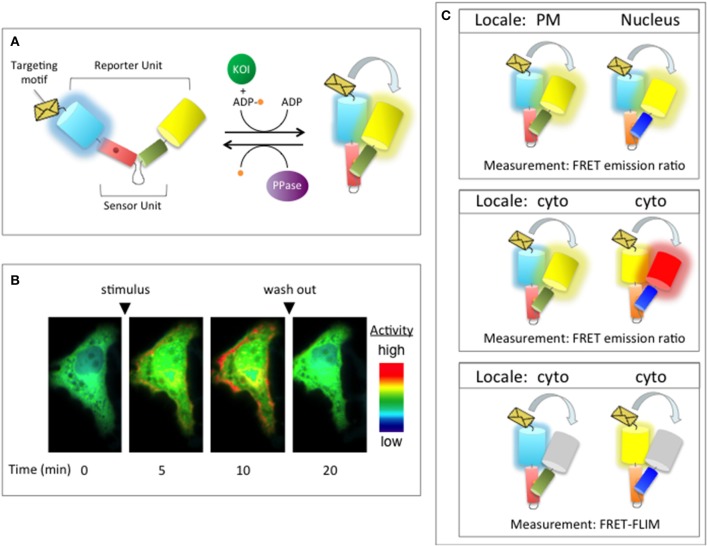
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources