

Whole exome sequence analysis of Peters anomaly
- PMID: 25182519
- PMCID: PMC4395516
- DOI: 10.1007/s00439-014-1481-x
Whole exome sequence analysis of Peters anomaly
Abstract
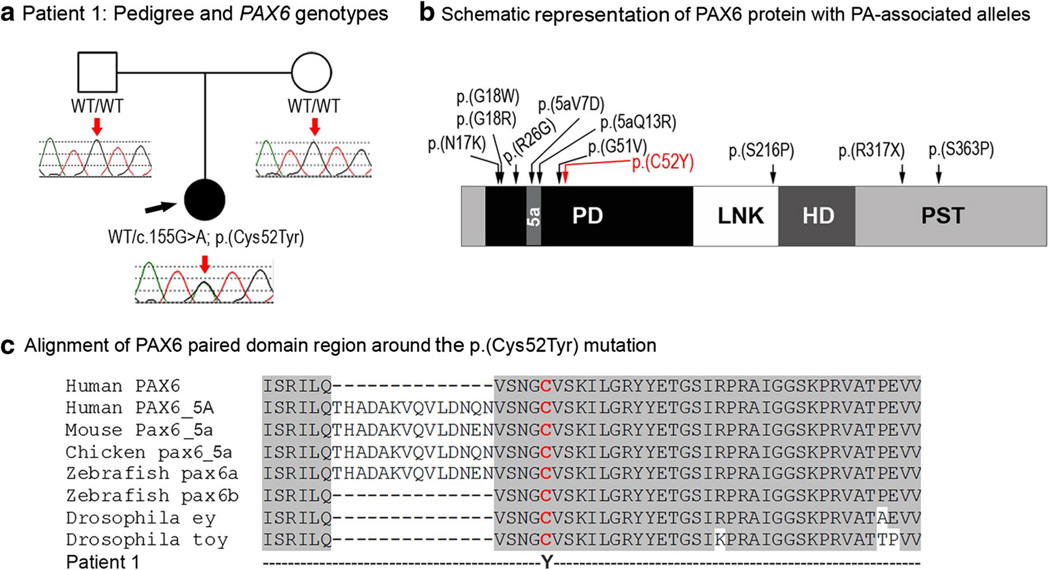
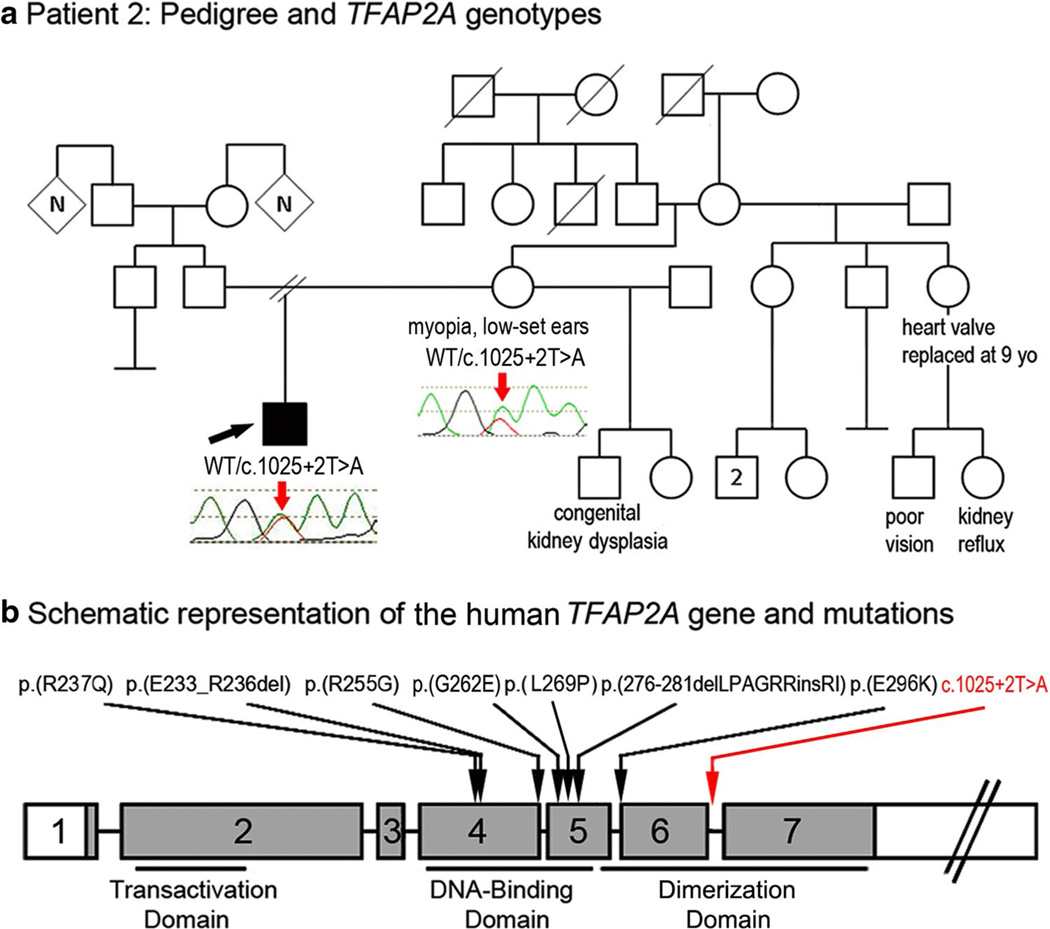
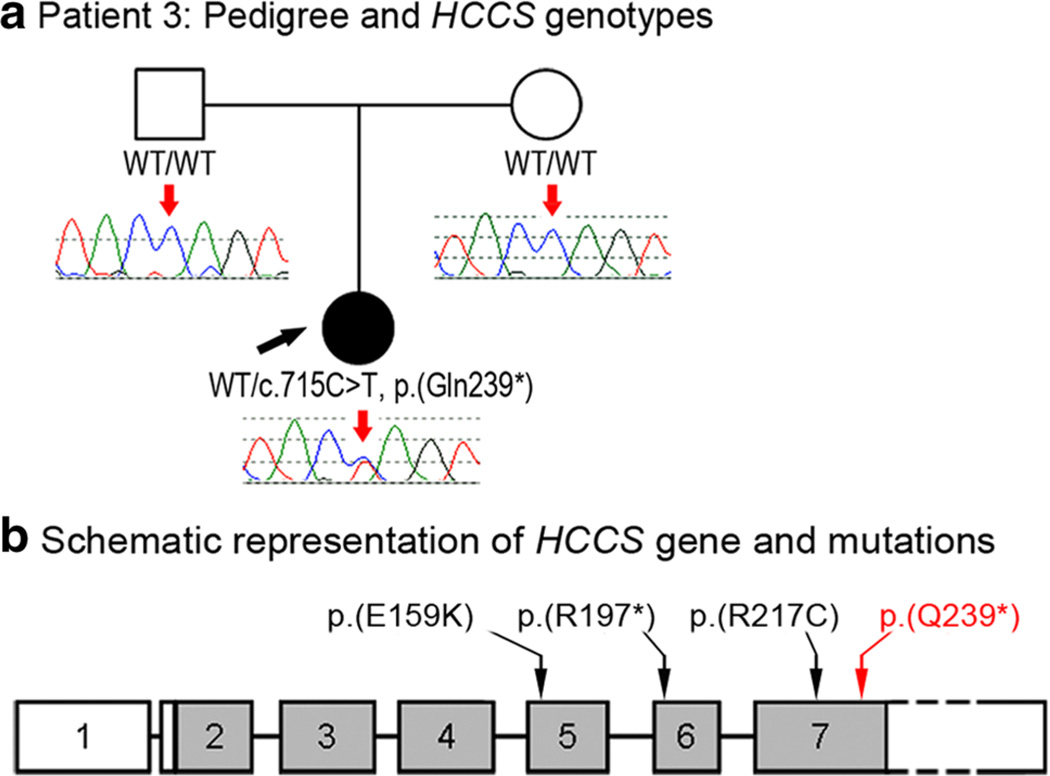
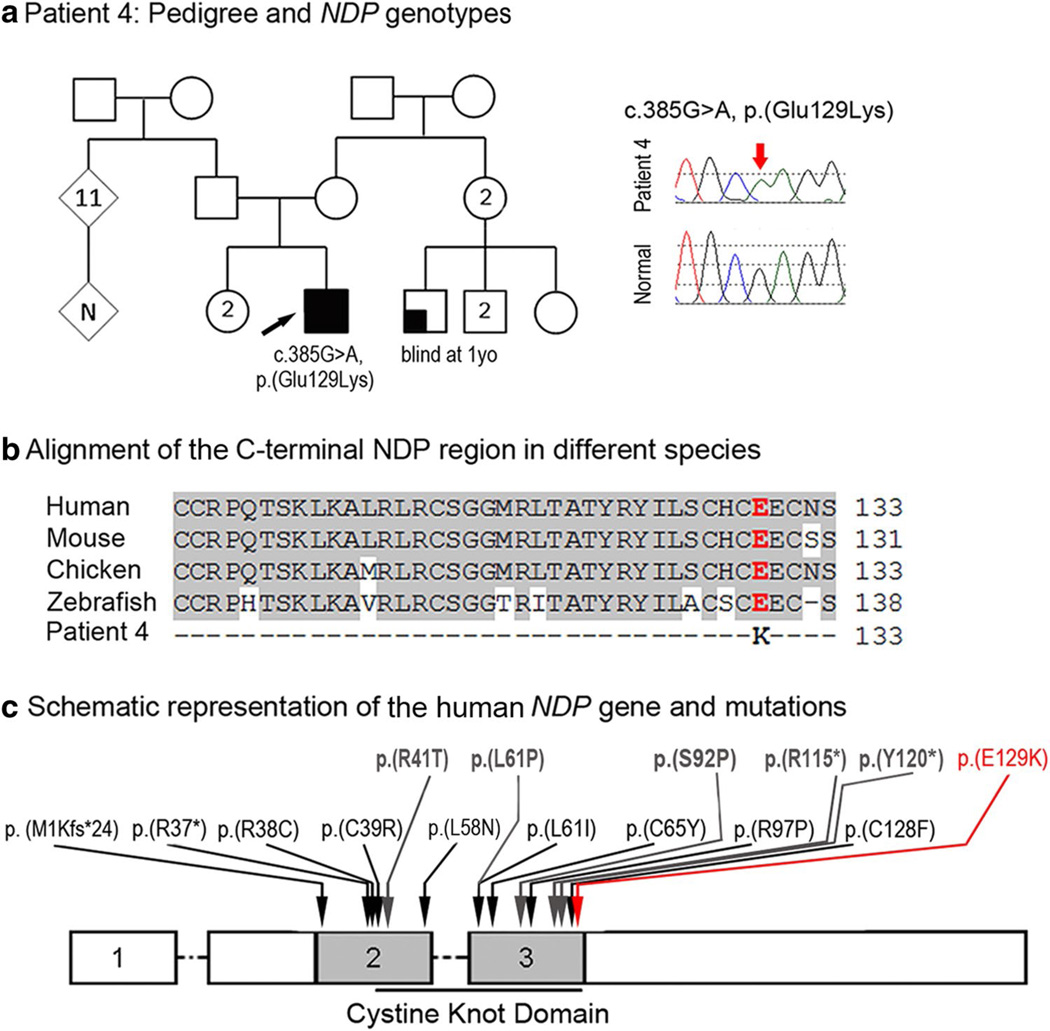
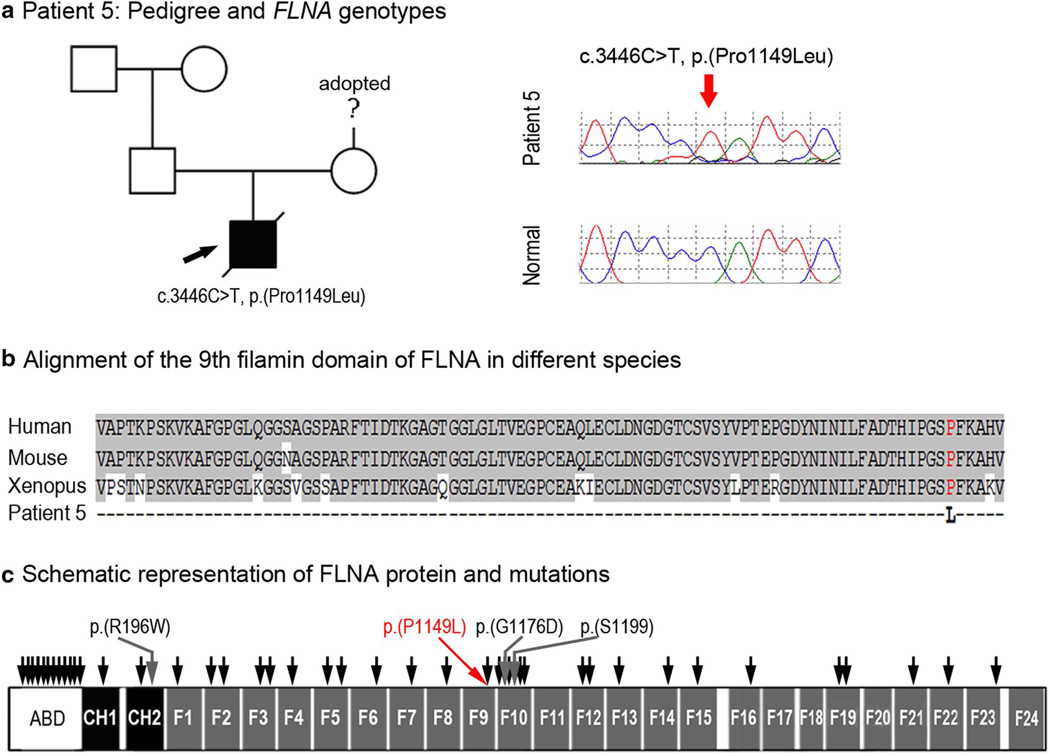
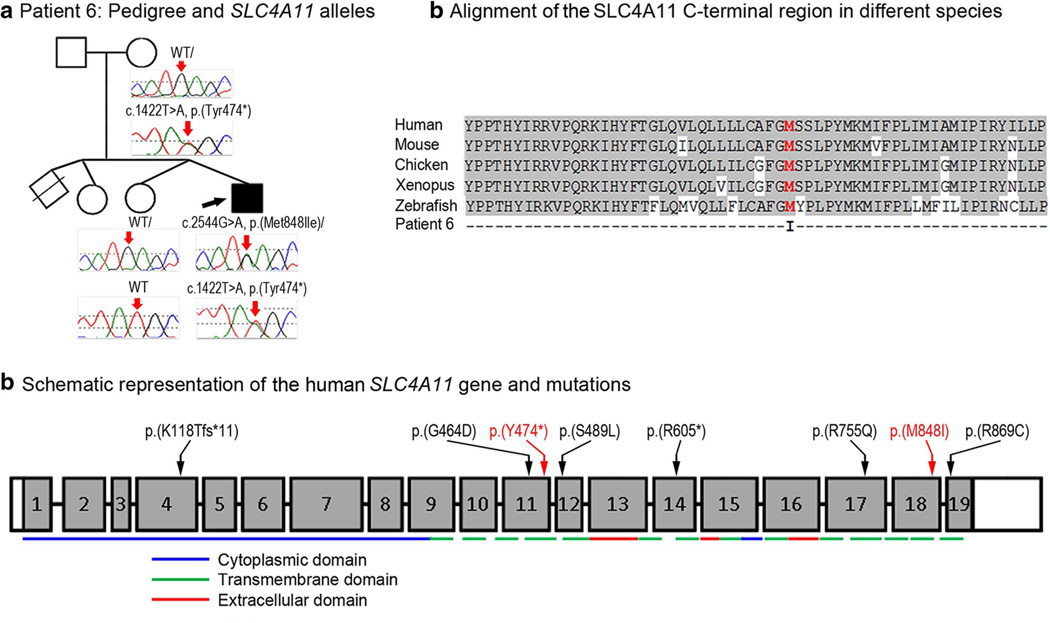
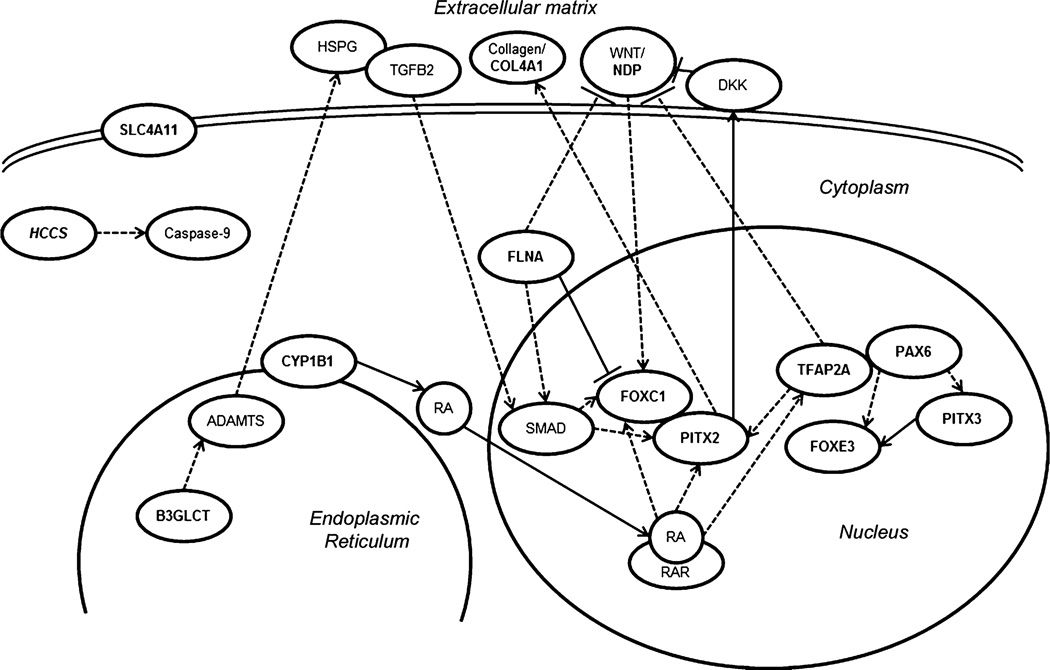
Peters anomaly is a rare form of anterior segment ocular dysgenesis, which can also be associated with additional systemic defects. At this time, the majority of cases of Peters anomaly lack a genetic diagnosis. We performed whole exome sequencing of 27 patients with syndromic or isolated Peters anomaly to search for pathogenic mutations in currently known ocular genes. Among the eight previously recognized Peters anomaly genes, we identified a de novo missense mutation in PAX6, c.155G>A, p.(Cys52Tyr), in one patient. Analysis of 691 additional genes currently associated with a different ocular phenotype identified a heterozygous splicing mutation c.1025+2T>A in TFAP2A, a de novo heterozygous nonsense mutation c.715C>T, p.(Gln239*) in HCCS, a hemizygous mutation c.385G>A, p.(Glu129Lys) in NDP, a hemizygous mutation c.3446C>T, p.(Pro1149Leu) in FLNA, and compound heterozygous mutations c.1422T>A, p.(Tyr474*) and c.2544G>A, p.(Met848Ile) in SLC4A11; all mutations, except for the FLNA and SLC4A11 c.2544G>A alleles, are novel. This is the first study to use whole exome sequencing to discern the genetic etiology of a large cohort of patients with syndromic or isolated Peters anomaly. We report five new genes associated with this condition and suggest screening of TFAP2A and FLNA in patients with Peters anomaly and relevant syndromic features and HCCS, NDP and SLC4A11 in patients with isolated Peters anomaly.
Conflict of interest statement
Figures
References
-
- Acharya M, Huang L, Fleisch VC, Allison WT, Walter MA. A complex regulatory network of transcription factors critical for ocular development and disease. Hum Mol Genet. 2011;20:1610–1624. - PubMed
-
- Ahmad N, Aslam M, Muenster D, Horsch M, Khan MA, Carlsson P, Beckers J, Graw J. Pitx3 directly regulates Foxe3 during early lens development. Int J Dev Biol. 2013;57:741–751. - PubMed
-
- Bamforth SD, Bragança J, Farthing CR, Schneider JE, Broadbent C, Michell AC, Clarke K, Neubauer S, Norris D, Brown NA, Anderson RH, Bhattacharya S. Cited2 controls left-right patterning and heart development through a Nodal-Pitx2c pathway. Nat Genet. 2004;36:1189–1196. - PubMed
-
- Baratz KH, Tosakulwong N, Ryu E, Brown WL, Branham K, Chen W, Tran KD, Schmid-Kubista KE, Heckenlively JR, Swaroop A, Abecasis G, Bailey KR, Edwards AO. E2-2 protein and Fuchs’s corneal dystrophy. N Engl J Med. 2010;363:1016–1024. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
